

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
基于图注意力网络和简单循环单元的化合物-蛋白质交互预测
[李淑红1  , 贾琳
, 贾琳1 ]
, 贾琳]
|
|



作者简介:

贾琳,硕士研究生,主要研究方向为深度学习.E-mail:568334521@qq.com.
现有基于深度学习的化合物-蛋白质交互预测方法未考虑数据的内部协变量偏移及序列数据的长距离依赖.针对此问题,文中提出基于图注意力网络和简单循环单元的化合物-蛋白质交互预测方法.利用图注意力网络-门控循环单元学习化合物分子的图级表示,利用多层简单循环单元学习氨基酸子序列的特征向量表示,结合多层前馈网络预测化合物-蛋白质的交互作用.实验表明,文中方法在2个公开数据集上的各项评估指标都有所提升,由此验证方法的有效性.
About Author:
JIA Lin, master student. Her research interests include deep learning.
The internal covariant shift of the data and the long distance dependence of the sequence data are not taken into account in the existing deep learning based compound protein interaction prediction methods. To solve the problem, a method for compound-protein interaction prediction based on graph attention network and simple recurrent unit is proposed. The graph attention network-gated recurrent unit is introduced to learn the graph-level representation of compound molecules, the multi-layer-simple recurrent unit is employed to learn feature vector representation of amino acid subsequences, and multilayer-feed-forward network is utilized to predict compound-protein interactions. Experiments show that the evaluation indexes of the proposed method are improved on 2 public datasets, and the effectiveness of the proposed method is verified.
本文责任编委 周水庚
Recommended by Associate Editor ZHOU Shuigeng
随着社会的发展, 多种新型疾病不断产生, 对人类生命健康造成巨大危害, 而药物发现是发现新药以治疗或治愈人类疾病的过程.药物靶标的识别是药物发现的重要环节, 其中一项关键任务是化合物-蛋白质相互作用(Compound-Protein Interactions, CPIs)的预测研究, 如何通过识别CPIs发现新药已成为研究人员关注的热点.CPIs预测具有广泛应用, 在预测药物靶向、探寻药物药效、预测药物副作用[1]及毒性等方面起到关键性作用.此外, 通过预测未知的CPIs也可用于药物重定位[2].
现有的CPIs预测方法可分为3类:基于生物化学的实验方法[3, 4, 5, 6, 7]、基于结构的计算方法[8, 9, 10, 11]、基于机器学习的方法[12, 13, 14].
基于生物化学的实验方法包括:宗可昕等[3]通过动力学实验原理检测药物中化合物对标靶蛋白质的结合亲和力.孙国章等[4]使用基于光谱的方法研究CPIs问题.Vilar等[5]提出基于3D化学相似性、靶标和不良反应数据的整合方法, 确定药物-蛋白质和药物不良反应之间的关联.Molina等[6]提出细胞热转变分析的实验方法.张建锋[7]使用电化学方法研究CPIs.
基于结构的计算方法包括:李玲等[8]通过分子对接的方法, 筛选能与靶蛋白有效结合的药物小分子化合物.Ma等[9]提出基于结构的虚拟筛选, 进行药物重新定位.贾聪敏[10]提出基于分子振动特征的药物靶点识别方法.Chow等[11]提出基于分子动力学的计算方法, 可计算化合物分子与相应蛋白质的结合能力, 从而计算二者的相互作用模式, 准确率较高, 但需要大量的专业知识, 即算法知识.
基于机器学习的方法包括:Jacob等[12]利用化学亚结构和蛋白质家族之间的张量积的特征, 并结合支持向量机(Support Vector Machine, SVM), 提出改进的化学基因组学方法.Bleakley等[13]提出二分图局部模型法(Bipartite Graph Local Model Method, BLM), 利用化学结构和蛋白质序列之间的相似性度量, 基于已知的交互训练SVM进行CPIs的预测.Coelho等[14]提出基于集成学习的方法, 结合随机森林与支持向量, 预测化合物蛋白质的交互.
虽然传统的基于机器学习方法应用于CPIs的预测具有一定效果, 但是传统机器学习方法属于浅层学习, 需要大量人工参与特征提取, 对于海量数据存在很大的局限性.为了解决此问题, Tian等[15]提出利用深度神经网络(Deep Neural Network, DNN)学习化合物-蛋白质对的特征, 预测性能较优.Nguyen等[16]提出使用图神经网络预测药物-靶标亲和力(Predicting Drug-Target Binding Affinity with Graph Neural Networks, GraphDTA).Tsubaki等[17]结合图卷积网络(Graph Convolutional Network, GCN)[18]与卷积神经网络(Convolutional Neural Networks, CNN)[19], 使用GCN处理化合物分子图, CNN处理蛋白质序列, 从而进行CPIs预测, 取得较优结果, 但预测性能和稳定性仍有进一步提升空间.因为GCN在处理图数据时需要访问整个图, 随着网络深度增加, 节点向量的表示会逐渐趋同, 导致节点的区分性变差.另外由于CNN的卷积核大小固定, 只能捕获滑动窗口内的子序列关系, 难以获取蛋白质中所有氨基酸子序列的依赖关系.
为了改善上述方法的泛化能力及稳定性, 本文提出基于图注意力网络和简单循环单元的化合物-蛋白质交互预测方法.利用图注意力网络[20]-门控循环单元[21](Graph Attention Network-Gated Recu-rrent Unit, GAT-GRU)学习化合物的潜在特征, 利用多层简单循环单元(Multi-layer-Simple Recurrent Units, ML-SRU)[22]学习蛋白质的氨基酸序列特征表示.再将学习到的化合物与蛋白质的特征向量表示进行拼接, 输入多层前馈网络以预测化合物-蛋白质的交互作用.实验表明, 本文方法在2个公开的CPIs数据集上的分类效果较优.
化合物-蛋白质对的交互预测任务旨在给出一个化合物分子和一个蛋白质的氨基酸序列, 判断二者是否交互, 实际上是一个二分类问题.形式化定义如下:给定一个化合物分子V=[v1, v2, …, vN]; 一个氨基酸序列S=[x1, x2, …, xM]; 交互的标签集合L={l1, l2}, 其中, l1表示交互的类别标签, l2表示不交互的类别标签.化合物-蛋白质对的交互预测任务是学习一个分类模型G:(V, S)→ L.模型输入化合物分子V和氨基酸序列S, 输出类别标签.
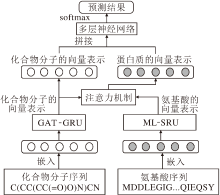
本文方法的整体流程如图1所示.主要过程如下.1)基于GAT-GRU的化合物分子的表示学习, 实现对化合物分子的特征向量化的表示.首先对化合物序列进行嵌入, 再利用图注意力层[20]实现对化合物分子特征的提取与表示, 并利用门控循环单元[21]实现对高阶邻居节点的特征进行抽取与过滤.2)基于ML-SRU的氨基酸序列的特征提取.首先对氨基酸序列进行嵌入保持与化合物的维度一致, 再利用多层简单循环单元提取氨基酸序列的特征并进行向量化表示.3)基于注意力机制的蛋白质向量表示, 拼接前两部分生成的向量表示, 作为网络输入, 通过注意力机制模拟相互作用强度, 形成蛋白质的向量表示, 送入多层前馈网络进行分类预测.
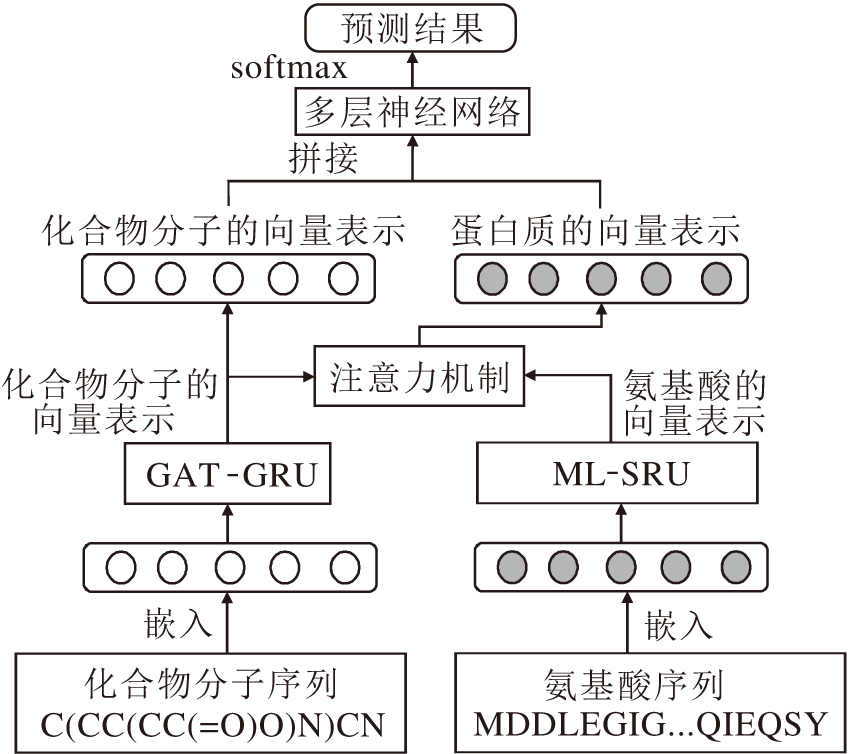 | 图1 本文方法流程图Fig.1 Flow chart of the proposed method |
基于GAT-GRU的化合物分子的表示学习模型主要由3个相同的基本模块堆叠而成, 每个模块由图注意力层-标准化-拼接-门控循环单元构成, 将各模块之间的计算结果逐层拼接以整合各模块的特征向量表示, 实现对高阶邻居节点的特征抽取与更新.另外, 考虑到数据经过图注意力卷积操作后其概率分布可能会发生偏移, 导致模型表达能力下降, 因此在进行图注意力卷积操作后引入实例标准化操作(Instance Normalization, IN)[23], 使新的分布更切合数据的真实分布, 保证模型的非线性表达能力.模型框架如图2所示.
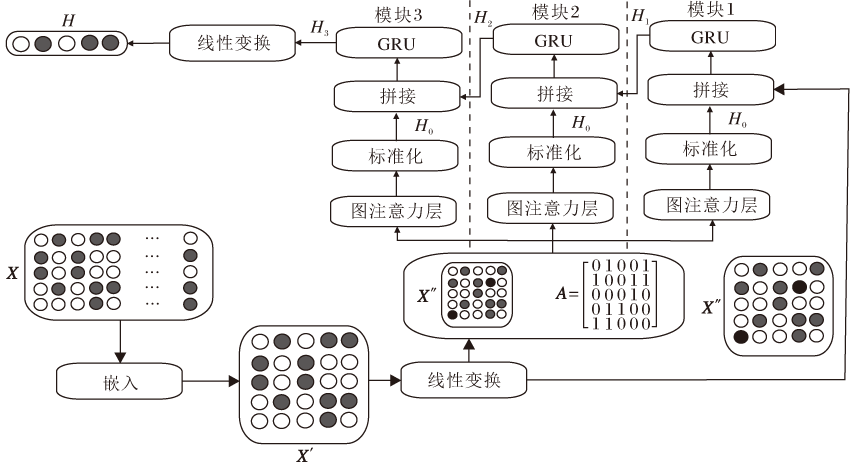 | 图2 基于GAT-GRU的化合物分子的表示学习模型框架Fig.2 Framework of representation learning model for compound molecules based on GAT-GRU |
由于网络输入是基于SMILES格式的化合物, 因此使用RdKit软件包将基于SMILES格式的数据转换为图结构的数据, 将化合物中的原子视为图中的节点, 将原子之间的化学键视为图中的边并对其进行建模.利用Rdkit提取相应原子特征信息, 存放于节点特征矩阵X∈ RN× C中, 原子间的结构信息存放于邻接矩阵A∈ RN× N中, X和A能为模型提供每个原子及其相邻原子的特征信息及结构信息.
节点特征矩阵X=[xij]N× C, 其中, N为节点数, C为特征向量的维度, X的第i行元素xi· 表示第i个原子vi对应的特征向量, X的第j列元素x· j表示各个原子的第j维特征.邻接矩阵A=[aij]N× N描述原子之间的结构关系, aij表示原子vi与vj的连接关系, 当原子vi、vj之间有键连接时, aij=1, 无键连接时, aij=0.
具体实现过程如下.
1)对X进行嵌入操作, 转换为低维向量表示形式X'∈ RN× C', C'为目标嵌入维度.为了增强节点特征向量的表征能力, 对X'进行线性变换, 即通过线性层学习权重矩阵W1∈ RC'× C', 获得新的节点特征矩阵X″=X'W1.
2)将X″和A一起送入各模块的图注意力层进行图注意力卷积操作, 学习化合物分子图中潜在的特征表示, 经标准化操作后得到特征表示H0∈ RN× C'.
3)将H0与未经过图注意力层处理的特征表示X″进行拼接, 送入门控循环单元进行进一步的特征提取与过滤, 得到特征表示H1∈ RN× C'.
4)将H1与模块2中以同样方式计算得到的H0进行拼接, 送入模块2的门控循环单元进行处理.重复此过程, 可依次得到H2∈ RN× C', H3∈ RN× C', 并对H3进行线性变换, 使H=H3W2, 其中, W2∈ RC'× C', H∈ RN× C'.此时H已融合各模块的特征信息, 将H按照特征维度对所有原子在该维度的特征值求平均, 获得该化合物分子的特征向量表示H∈ R1× C'.
具体公式如下:
H0=norm(GATConv(Linear(x), A)),
H1=GRU(concat(H0, Linear(x))),
H2=GRU(concat(H0, H1)),
H3=GRU(concat(H0, H2)),
H=mean(Linear(H3)),
其中, H0表示由图注意力层和标准化操作得到的隐藏表示, Hi(i=1, 2, 3)表示各个模块计算的隐藏特征表示, norm表示标准化操作, GATConv表示图注意力层, GRU表示门控循环单元, concat表示拼接操作, Linear表示线性变换, mean表示对各维度的特征表示求均值, H表示最后得出的化合物分子的特征向量表示.
具体地, GAT使用自注意力机制解决基于图卷积模型的不足.通过注意力机制对化合物分子图中的相邻节点进行图卷积操作, 将相应的权重分配给不同的邻居节点, 并对邻域内节点特征进行加权求和, 充分考虑相邻节点间的结构关系, 将图中各节点特征之间的相关性融入模型中, 有助于模型学习结构信息.GATConv操作利用图的结构信息A和节点特征信息X进行节点向量表示, 将待更新的节点记为vi, 对应的特征向量记为hi, 经注意力机制的聚合操作输出新的特征向量表示h'i, 计算每对节点vi和vj间的权重系数:
eij=LeakyReLU(aT(Whi‖ Whj)),
其中, a为节点特征的参数, W为特征变换的权重.为了对不同邻居节点分配相应权重, 使用softmax对权重进行归一化操作, 归一化后权重系数为
α ij=softmax(eij)=
其中N(vi)表示节点i的邻居节点, 则节点vi更新后的特征向量

给定氨基酸序列S=[x1, x2, …, xM], 首先利用n-gram方法划分给定的氨基酸序列, 本文使用n=3的划分, 形成连续且重叠的子序列:
[x1x2x3]=s1,
[x2x3x4]=s2,
︙
[xM-2xM-1xM]=sM-2.
在数据处理时将氨基酸的字符序列逐一映射为按其长度排序的索引形成字典, 按照字典对每个氨基酸子序列进行向量化表示.将划分后的字符序列S'=[s1, s2, …, sM-2]按照字典映射为数值形式的向量Q=[q1, q2, …, qM-2], 其中qi表示第i个氨基酸序列si的向量形式.
多数现有方法使用CNN处理氨基酸序列, 但是CNN在处理长距离信息提取时存在困难, 因此考虑使用循环神经网络(Recurrent Neural Network, RNN)代替CNN, 而RNN在处理长序列时存在长距离依赖问题, 原因在于其串行的计算方式容易产生梯度消失或梯度爆炸.长短期记忆网络(Long Short-Term Memory, LSTM)和门控循环单元(Gated Recurrent Units, GRU)以门控方式控制信息传递, 各个门及当前隐状态的计算依赖于上一步隐藏状态的输出.为了降低对门控单元和隐藏状态计算的复杂程度, 并且能在长序列中并行计算, 对简单循环单元(Simple Recurrent Units, SRU)[22]进行改进, 使各个门和隐状态的计算仅与当前输入有关, 不依赖于上一步的隐藏状态.另外, 在SRU的高速网络中引入跳跃连接, 使梯度直接向前传递, 使模型即便叠加多层, 仍能避免梯度消失并保持模型性能, 因此本文使用堆叠的多层SRU进行特征提取.
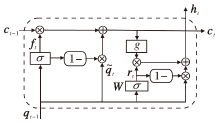
基于ML-SRU的氨基酸序列的特征提取模块的具体过程如下.将氨基酸序列嵌入实值向量空间中, 输入至堆叠的ML-SRU中进行特征提取.SRU处理特征信息的过程为t时刻, 读取输入向量qt, 计算细胞状态序列:
ct=ft☉ct-1+(1-ft)☉
并捕获序列的信息, 其中,
ft=σ (Wf
表示遗忘门用于控制输入信息
rt=σ (Wr
用于将当前的输入信息qt及当前时刻的细胞状态ct进行组合, 输入前一时刻细胞的平均状态ct-1及当前输入信息
ht=rt☉g(ct)+(1-rt)☉qt,
其中, W、Wf、Wb、bf、br为可训练的参数矩阵.由于
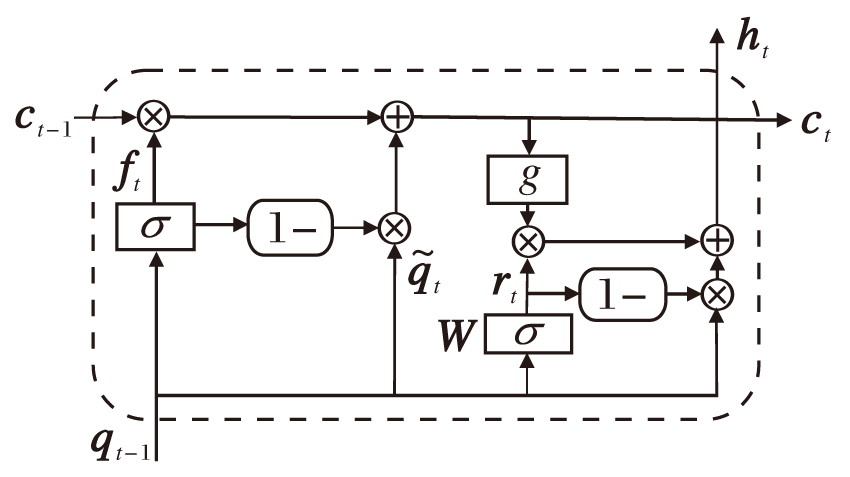 | 图3 基于ML-SRU的氨基酸序列的特征提取框架Fig.3 Feature extraction framework of amino acid sequence based on ML-SRU |
在经过前2个模型的特征提取后, 得到化合物分子的向量表示及氨基酸子序列的向量表示.由于不同的氨基酸子序列与化合物分子的作用强度存在差异, 在计算时需要考虑二者间的相互作用, 因此考虑使用软性注意机制[24]模拟这种相互作用.注意力机制通过对数据进行加权处理, 突出显示更重要的特征信息.本文使用注意力机制学习各氨基酸片段的重要性, 即某个氨基酸子序列对化合物越重要, 则赋予该子序列越大的权重.使用注意力的分数表示相互作用的强度, 通过对氨基酸序列的加权求和得到蛋白质的向量表示.
给定经ML-SRU特征提取后蛋白质的氨基酸子序列的一组隐藏向量Q=[q1, q2, …, qM-2], 蛋白质向量的表示过程如下所示:
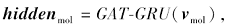
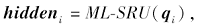


其中:vmol表示输入GAT-GRU的化合物分子的向量表示; qi表示输入ML-SRU中的第i个氨基酸子序列; hiddenmol表示经过GAT-GRU处理后的化合物分子的隐藏向量表示, hiddeni表示经过ML-SRU处理后得到的第i个氨基酸子序列的隐藏表示, 将基于hiddenmol和hiddeni的点积计算的α i作为注意力分数, 通过对hiddeni的加权求和计算得出最终的蛋白质序列的隐藏表示; W表示权重矩阵, b表示偏置; GAT-GRU(· )表示GAT-GRU模型, ML-SRU(· )表示ML-SRU模型.注意力分数是通过在网络结构中加入一个前馈神经网络进行学习的.使用神经注意机制, 可反映出在相互作用中蛋白质的哪些区域是重要区域.
将得到的蛋白质的向量表示与化合物分子的向量表示进行拼接, 输入多层前馈网络, 最后一层网络得到的特征向量可作为分类器所需的特征, 经过softmax激活函数进行激活, 输出化合物-蛋白质对交互的概率:
pk= softmax(Woutconcat(hiddenmol, hiddeni)+bout).
其中, Wout表示全连接层的权重矩阵, bout表示全连接层的偏置.模型使用有监督学习的方式进行学习, 使用反向传播算法更新模型参数, 并利用最小化交叉熵训练模型.为了防止过拟合在交叉熵公式后加上L2正则项, 交叉熵损失函数表示如下:
Loss=-
其中:k=0表示无交互, k=1表示交互; n为训练集的数据量;
实验使用Human和C.elegans[25] 2个生物领域常用的基准CPIs数据集进行评估, 具体数据集信息如表1所示.
| 表1 实验数据集 Table 1 Experimental datasets |
每个数据集中记录化合物与蛋白质之间有无交互的关系, 每个化合物以SMILES格式表示, 每个蛋白质以氨基酸序列的形式表示, 数据样本中的标签分为有交互与无交互, 分别用1和0表示.
实验中每个数据集均按照80%、10%、10%的比例划分为训练集、验证集、测试集.
超参数设置如下:使用Adam优化器进行训练, 学习率设置为1e-4, 权重衰减设置为1e-6, 学习率衰减为0.9, 嵌入维度为30, 输出层层数及GAT-GRU中的模块数均为3, SRU层数选择为5, 迭代次数为100轮.
实验环境设置如下.操作系统为Windows10, 内存8.0 GB, 处理器Intel(R)Core(TM)i5-5350U CPU@1.8 GHz, 使用的框架是Pytorch, 编程语言为Python.
为了衡量方法性能, 采用如下评价指标, 包括曲线下面积(Area under Curve, AUC), 精确率(Preci-sion), 召回率(Recall), F1值:
Precision=
Recall=
F1=
其中:TP表示真正例, 实际为正且预测为正; FP表示假正例, 实际为负但预测为正; FN表示假反例, 实际为正但预测为负; TN表示真反例, 实际为负且预测为负.F1值用于衡量Precision和Recall, 是这两个值的调和均值, 综合反映整体情况.
在泛化能力的实验中使用正确率(Accuracy, ACC)作为评估标准:
Accuracy=
为了验证本文方法预测的有效性, 选取目前最具代表性的2种基于深度学习[16, 17]方法和6种基于机器学习[25]的方法进行实验对比.为了准确复现基准方法, 部分对比实验使用原论文设置的相关参数并在相同数据集上运行该论文中开源代码, 将得到的结果作为实验对比依据.
用于对比的2种基于深度学习方法如下.
1)GraphDTA[16].基于GNN和CNN的组合, 结合池化操作, 用于计算药物-靶标结合亲和力, 是一种回归任务, 实验时对其进行微调, 改为二分类问题, 用于CPIs预测.
2)GCN-CNN[17].基于GCN和CNN的组合, 利用GCN提取化合物分子特征, CNN提取蛋白质序列特征, 并利用注意力机制模拟二者相互作用, 用于CPIs的预测.
用于对比的6个机器学习方法如下.
1)朴素贝叶斯(Naive Bayesian, NB).
2)k近邻算法(k-Nearest Neighbor, KNN).
3)随机森林(RandomForest, RF).
4)L1(L1-logistic).
5)L2(L2-logistic).
6)SVM.
实验设置如下:NB、KNN、RF由Weka3.7运行, L1、L2由liblinear1.94运行, SVM由libsvm3.17运行.各分类方法分别在各软件的默认环境下进行.
本文方法和基于深度学习方法在2个数据集上的性能对比如表2所示.由表可看出, 本文方法的预测结果更优.在Human数据集上, 相比GraphDTA、GCN-CNN, 本文方法的AUC值分别提升3.5%和2.7%, F1值分别提升7.9%和6.5%.在C.elegans数据集上, 相比GraphDTA、GCN-CNN, 本文方法的AUC值分别提升2.2%和1.6%, F1值分别提升5.2%和3.9%, 从而验证本文方法的有效性.
| 表2 3种方法在2个数据集上的性能对比 Table 2 Performance comparison of 3 methods on 2 datasets |
本文方法与机器学习方法的性能对比如表3所示.
| 表3 7种方法在2个数据集上的性能对比 Table3 Performance comparison of 7 methods on 2 datasets |
由表3可看出, 相比表现最优的RF, 本文方法在Human数据集上AUC值提升5.7%, 在C.elegans数据集上AUC值提升3.2%.这可能是因为本文方法属于端到端的表示学习方法, 可基于数据进行表征学习, 自动学习到有意义的数据特征信息, 从而反映化合物-蛋白质交互性质.而上述机器学习方法以化合物的结构和蛋白质的结构作为输入特征, 利用化合物分子图和蛋白质序列的生物化学特性, 分类效果的优劣很大程度上依赖于人工设计的特征.
为了验证本文方法中各模块的有效性, 进行消融实验.对比方法如下.
1)GCN-ML-SRU.将本文方法中的GAT-GRU模块替换成普通GCN网络, 并与ML-SRU结合.
2)GAT-ML-SRU.将本文方法中的GAT-GRU模块替换成普通GAT网络, 并与ML-SRU相结合.
3)None-attn.在本文方法的基础上在计算蛋白质向量的过程中取消注意力机制, 直接将SRU特征提取后的特征向量作为蛋白质向量表示.
各方法的消融实验结果如表4所示.由表可见, 对比GAT-ML-SRU, 本文方法在Human、C.elegans数据集上的AUC值分别提升1.3%和0.6%, 表示在图注意力卷积操作中引入GRU单元有利于进一步提取高阶邻域信息, 从而验证GRU单元的有效性.对比GAT-ML-SRU、GCN-ML-SRU可发现, GAT-ML-SRU比GCN-ML-SRU的实验结果更优, 表示基于GAT的方法提取节点特征信息的能力更强, 而SRU能缓解CNN难以捕获长距离特征信息问题.对比本文方法与None-attn发现, 当不使用注意力对蛋白质进行表示时, 方法预测性能的评价指标均呈现不同程度的下降, 表示使用注意力机制能有效捕获蛋白质的特征信息, 说明蛋白质向量表示的改进可使方法性能得到提升.
| 表4 各方法在2个数据集上的消融实验结果 Table 4 Ablation experiments of different methods on 2 datasets |
本节分析可能对CPIs预测性能产生影响的因素, 主要从SRU网络层数、嵌入维度、是否使用标准化操作及GAT-GRU中的模块数进行分析.
通常多层神经网络的非线性表征能力较强, 能学习到更复杂的变换, 可拟合更高级的特征, 使网络性能有所改进.SRU网络层数对评价指标的影响如图4所示.由图可看出, 本文方法在层数为5时表现最佳, 但随着层数的增加, 性能并不会一直提升.其原因可能是随着网络层数的增加, 参数增加, 训练难度增大, 导致通用性变差.
 | 图4 SRU的网络层数对评价指标的影响Fig.4 Effect of SRU network layers on evaluation metrics |
不同嵌入维度对评价指标的影响如图5所示.由图可看出, 嵌入维度为10时AUC值低于嵌入维度为30的AUC值, 当嵌入维度为30时效果最佳, 而嵌入维度为50的AUC值、F1值均有所下降.其原因可能是虽然嵌入维度越高能保留的特征信息也越多, 但引入的噪声也越多, 导致时间复杂度和空间复杂度也会随之升高.这在一定程度上表示CPIs预测问题适用于低维嵌入, 既能获取足够的特征信息, 也不会增加额外的时间和空间复杂度.
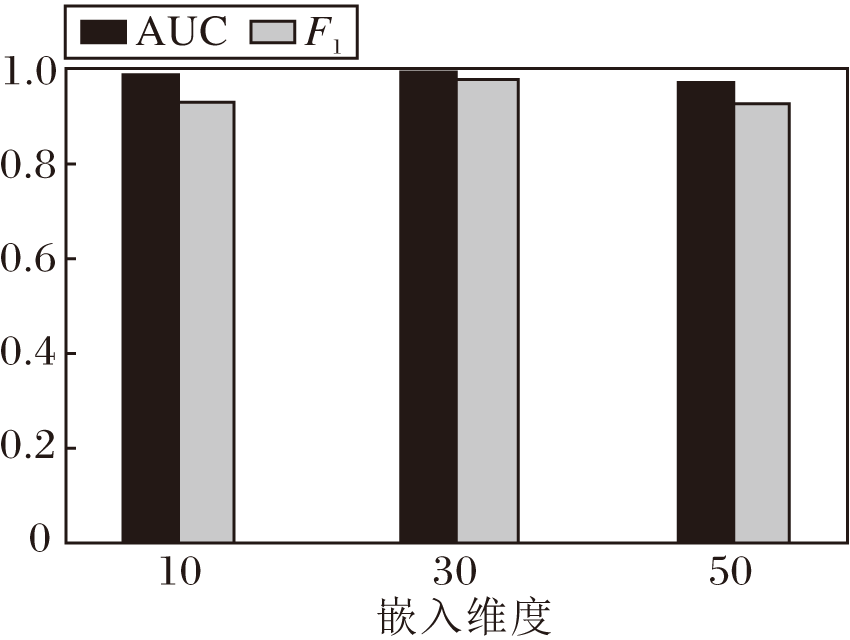 | 图5 不同嵌入维度对评价指标的影响Fig.5 Effect of different embedding dimensions on evaluation metrics |
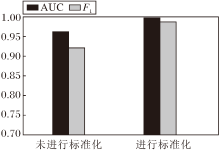
通常随着网络层数的加深, 内部协变量偏移问题可能会愈加严重, 因此是否使用特征向量标准化的模型对AUC值和F1值的影响如图6所示.由图可看出, 相比未进行标准化操作的GAT-GRU, 使用标准化操作的GAT-GRU的AUC值和F1值更高, 这可能是由于输入数据经过网络处理后, 概率分布会发生偏移, 导致数据的表征能力下降, 影响网络收敛.但是, 对其进行标准化操作能使经网络处理后的数据分布与输入数据的分布基本保持一致, 有助于方法收敛.
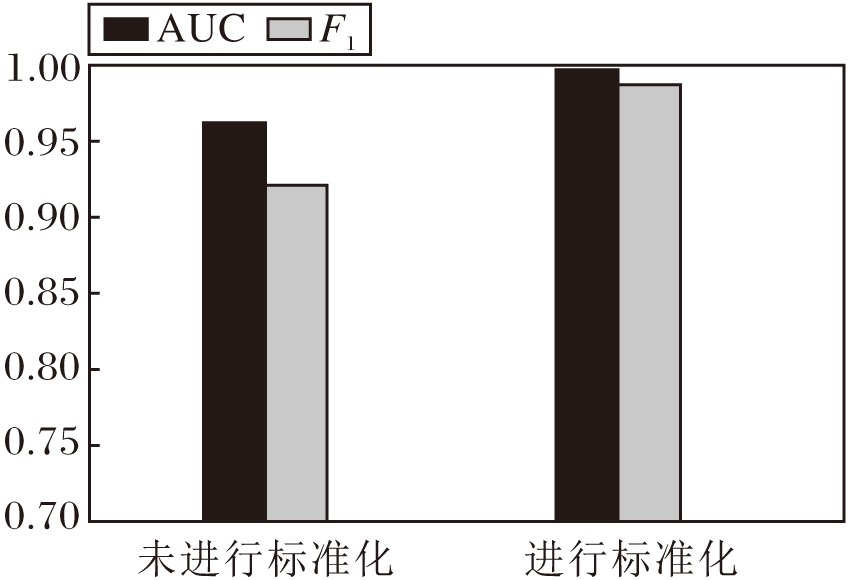 | 图6 特征向量标准化对评价指标的影响Fig.6 Effect of eigenvector normalization on evaluation metrics |
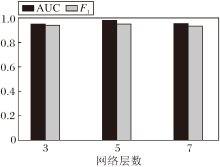
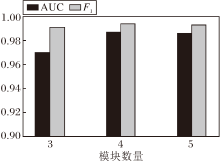
GAT-GRU中不同模块数对AUC值和F1值的影响如图7所示.由图可看出, 模块数量从3增加到4, F1值增加, AUC值并无大幅提升, 当模块数增加到5时, AUC值和F1值甚至略有下降.其原因可能是更深的网络带来的网络退化, 并且随着模块数的增加, 计算开销也会随之增加, 因此选择合适的模块数对实验结果至关重要.
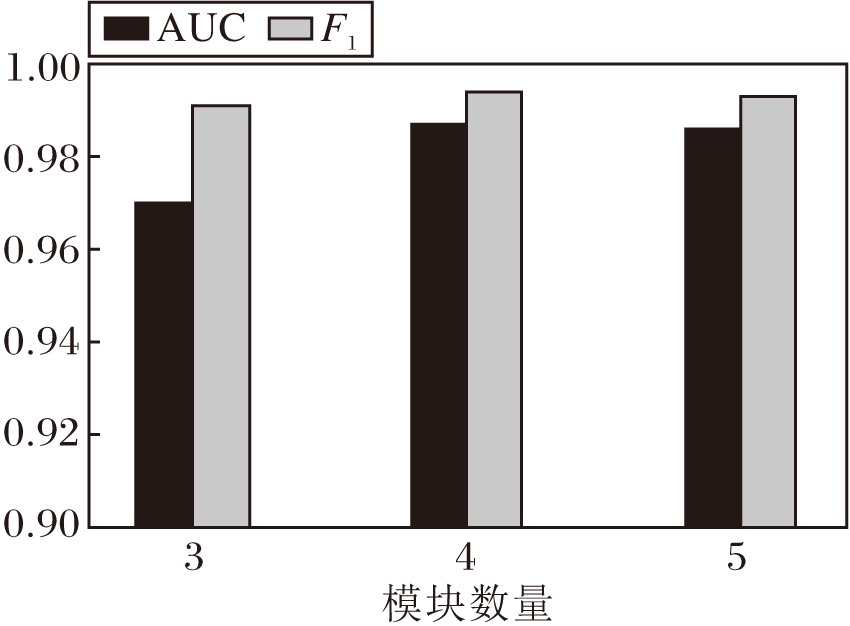 | 图7 不同模块数对评价指标的影响Fig.7 Effect of the number of different modules on evaluation metrics |
为了进一步验证本文方法的泛化能力, 在2个数据集上进行评估实验.设置数据集上80%的样本为训练集, 20%的样本为测试集.对比方法采用性能较优的GCN-CNN[16], 结果如表5所示.由表可看出, 本文方法略优于GCN-CNN, 泛化能力更优.从测试结果的方差可见, 本文方法的方差较小, 说明本文方法的性能较稳定.
| 表5 两种方法的泛化性能对比 Table 5 Generalized performance comparison of 2 methods |
在实际问题中, 药物发挥作用体现在小分子化合物与蛋白质活性位点发生交互, 使蛋白质的结构略有变化, 以此调节机体的生理活动.故不同位点相互作用强度有所不同, 通过使用注意机制可考虑到蛋白质中某些重要区域对交互的影响, 因此以得分较高的注意力权重对蛋白质的向量进行表示.
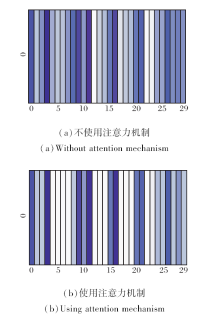
注意力机制对蛋白质向量表示的影响如图8所示, 由图可看出, 使用注意力机制的蛋白质向量表示对化合物与氨基酸相互作用越强, 位点颜色越深.通过减少对非重要位点的注意力, 突出显示蛋白质的重要区域, 达到增加模型准确率的效果.
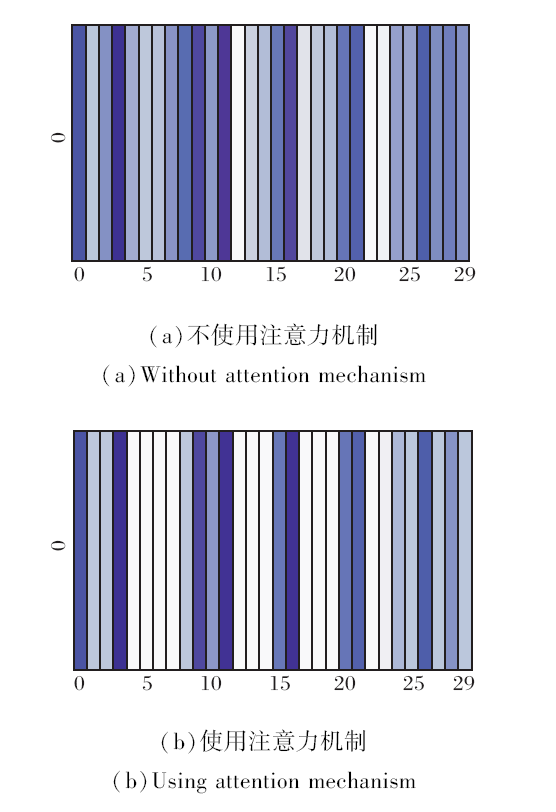 | 图8 是否使用注意力机制对蛋白质向量表示的影响Fig.8 Effect on protein vector representation using the attention mechanism or not |
CPIs是药物发现过程中一项关键的任务, 现有的基于深度学习的CPIs预测方法多采用图神经网络对化合物进行特征提取, 但未考虑数据的内部协变量偏移问题.有些方法采用传统卷积神经网络处理蛋白质序列, 但卷积神经网络难以捕获子序列间的关系, 导致模型表达能力较低.与此同时, 引入注意力机制的图神经网络能够自适应地提取重要节点的特征信息, 实例标准化操作可有效解决内部协变量偏移问题, 门控循环单元模型能对特征信息进行更新与过滤, 简单循环单元模型由于独特的结构, 能实现对数据的并行处理, 防止梯度消失.基于此种情况, 本文提出基于图注意力网络和简单循环单元的化合物-蛋白质交互预测方法.使用基于图神经网络与循环神经网络结合的方法对CPIs问题进行预测, 结合图注意力网络与门控循环单元, 提取化合物分子特征, 实现对高阶邻居节点的特征信息的自适应提取与更新.同时引入标准化操作, 使方法收敛更稳定, 并通过堆叠多层简单循环单元对蛋白质的氨基酸序列进行处理, 有效捕获子序列之间的信息.实验验证本文方法的有效性.今后将进一步利用基于Transformers的双向编码表征(Bidirectional En-coder Representations from Transformers, BERT)进行CPIs的预测, 提高方法性能.
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|