
{kind=link}
基于多尺度局部累积特征和神经网络的抗肿瘤药物反应预测
[韩睿1  , 郭成安
, 郭成安1 ]
, 郭成安]
|
|



作者简介:

韩 睿,博士研究生,主要研究方向为生物信息处理、医学智能.E-mail:tracyhan@mail.dlut.edu.cn.
目前已有的研究结果表明现有抗肿瘤药物的有效性高度依赖于患者的基因组学特征.如何为每位肿瘤患者量身定制最佳的治疗方案是重要又富有挑战性的前沿课题.针对该课题,文中提出抗肿瘤药物反应预测方法,运用机器学习技术,对患者肿瘤基因测序数据进行处理、特征提取及建模,预测各种不同抗肿瘤药物的疗效反应.首先,提出基于多尺度关联规则的数据挖掘方法,对基因组学数据进行不同尺度的特征挑选.进而通过累积窗函数对挑选后的基因组学数据进行局部累积,进一步执行数据压缩,提取具有较强整体表达性的基因特征信息.然后,以多层全连接神经网络为模型、以提取的多尺度累积基因特征为输入样本,进行训练和建模.最后,分别采用特征融合和决策融合,实现某一肿瘤基因测序数据对于各种不同抗肿瘤药物反应结果的预测.在COSMIC、GDSC数据库上的仿真实验表明,文中方法在敏感性、特异性、准确率、特性曲线面积值等关键性能指标上均取得较优值.
About Author:
HAN Rui, Ph.D. candidate. Her research interests include biological information processing and medical intelligence.
Medical research results show that the effectiveness of an antitumor drug is highly dependent on the genomic characteristics of patients. How to customize an optimal medical treatment for each tumor patient is an extremely important and challenging research topic. Aiming at this subject, a set of methods to predict the efficacy response of various antitumor drugs is proposed by machine learning technology for data processing, feature extraction and modeling of the tumor gene sequences of patients. Firstly, a data mining algorithm based on multiscale association rules is proposed for feature selection at different scales of genomics data. Then, the selected genomics data are locally accumulated by the cumulative window function to further compress the data and extract the gene feature information with stronger overall expression. Based on the above, a fully connected multi-layer neural network is designed and the extracted multiscale cumulative gene features are treated as input samples to train the network. Finally, two fusion methods, including feature fusion and decision fusion, are utilized to predict the responses of a tumor gene sequence to different antitumor drugs, respectively. Results of simulation experiments show that the proposed approach is superior in key performance indexes, such as sensitivity, specificity, accuracy and the area under characteristic curve.
本文责任编委 周水庚
Recommended by Associate Editor ZHOU Shuigeng
在精准肿瘤学研究领域, 研究人员目前正针对如何能为每位患者量身定制最佳的治疗方案进行深入探索[1].实施精准肿瘤学医疗技术的一个重要先决条件是能根据患者肿瘤的特定分子特征正确识别与所用药物反应相关的生物标记物.然而, 药物反应与肿瘤分子的生物特征之间的关系十分复杂, 目前尚未提出有效模型进行准确描述.当前的机器学习技术、不断积累的医疗大数据、以及将二者结合而开展的研究工作, 为解决这一问题提供一个有效途径.近年来, 随着个体基因测序技术的日益完善, 针对患者个人的肿瘤衍生分子数据, 对用于治疗肿瘤的药物活性进行无偏见、精确地建模与预测, 已成为实现肿瘤患者个体化、精准治疗的关键.因此, 该项研究不仅可促进精准肿瘤学的发展, 还能显著降低临床实施的成本, 使更多的患者受益.
目前, 一些国际医学研究机构已公开发布临床药物基因组学数据集, 为科研人员从事抗肿瘤药物反应预测研究提供重要的基础数据.2004年, 英国威康信托基金会桑格研究所发布COSMIC数据库[2].该数据库汇集人类肿瘤细胞系的基因突变信息, 其中一个专门组成部分CCLP(COSMIC Cell Lines Project)是对最常使用的1 000多种肿瘤细胞系的深入分析数据.2009年, 英国桑格研究院推出肿瘤药物敏感性基因组学GDSC(Genomics of Drug Sensitivity in Cancer)数据库[3], 提供1 000多种肿瘤细胞系的染色体拷贝数变异、基因表达等基因组学数据及265种抗肿瘤药物作用在肿瘤细胞系的半抑制浓度(Half Maximal Inhibitory Concentration), 即IC50值.
抗肿瘤药物反应预测研究大多基于高通量测序技术收集的不同基因组水平信息[4].相比上万维的基因组学特征, 肿瘤细胞系的数量通常只有一千个, 而在全部的基因组学特征中, 可能只有一部分与某一抗肿瘤药物反应相关.因此, 如何从原始的基因组学数据中提取合适的特征子集是预测抗肿瘤药物反应的关键.Xu等[5]提出AutoBorutaRF, 采用自编码器网络筛选一个适当维度的特征, 再利用Boruta算法进一步确定随机森林的输入特征.Vougas等[6]提出DLNNs(Deep Learning Neural Networks), 基于关联规则挖掘算法, 从基因表达、拷贝数变异和基因突变三种基因组学数据中提取单尺度的基因组学特征, 并从中随机挑选200维特征作为预测抗肿瘤药物反应的每个子分类器的输入特征.Sharifi-Noghabi等[7]采用3个编码子网络, 分别学习基因表达、基因突变、拷贝数变异三种基因组学数据的特征表示, 并将上述单组学特征连接成一个多组学特征, 作为预测子网络的输入特征.
用于抗肿瘤药物反应预测的机器学习方法主要包括:逻辑回归、弹性网络、随机森林、KNN(K-Nearest Neighbor), SVM(Support Vector Machine)及浅层神经网络等.近年来, 深度学习成为药物反应预测领域中一种值得研究的方法[8], 可分为两类.1)基于深度神经网络(Deep Neural Networks, DNN)的方法.Vougas等[6]将多个子分类器的输出加权平均后, 获得最终预测抗肿瘤药物反应的输出.每个子分类器均采用包含3个隐层的DNN结构.Sharifi-Noghabi等[7]提出MOLI(Multi-omics Late Integra-tion), 采用一个集成的多组学特征作为输入, 预测靶向药物家族的药物反应.其中的编码子网络和预测子网络均采用单隐层的DNN结构.Li等[9]提出DeepDSC(A Deep Learning Method to Predict Drug Sensitivity of Cancer Cell Lines), 将基因表达特征和药物的化学特征一起作为输入, 用于预测细胞系的药物敏感性, 预测网络采用包含4个隐层的DNN结构.Zhu等[10]提出迁移学习框架, 建立一般药物反应的预测模型, 采用7个隐层的DNN结构.2)基于深度卷积神经网络(Convolutional Neural Networks, CNN)的方法.Chang等[4]提出CDRscan(Cancer Drug Response Profile Scan), 实现对抗肿瘤药物的IC50值的预测, 其中2个输入CNN分支分别对细胞系的基因突变和药物的分子结构信息进行建模.在文献[4]的基础上, Bai等[11]进一步针对联合采用基因测序信息和药物分子结构信息的方法进行研究, 提出采用双输入和双输出的深度CNN, 实现对所用药物的敏感性及IC50值的预测, 取得较优效果.
虽然现有方法在解决抗肿瘤药物反应预测的关键问题上取得一定进展, 但仍存在如下问题.1)如何从原始的肿瘤细胞系提取合适的基因组学特征.Vougas等[6]基于关联规则挖掘, 提取单尺度的基因组学特征, 从中随机挑选多组特征子集分别作为待选特征, 虽然取得一定效果, 因其随机性较大, 难以覆盖全部基因组学特征, 所以无法保证具有稳定的性能.同时由于该方法需要采用多组待选特征分别进行训练, 需要的预测子网络数目较多, 造成训练耗时过长.另外, 采用单一规则提取基因组学特征, 可能会排除某些对预测模型有用的输入特征.2)在已有用于抗肿瘤药物反应研究的医学数据样本中, 正样本数据相对偏少, 对于训练过深的网络模型容易产生过拟合问题, 泛化能力严重下降.
针对上述的特征提取和预测模型设计等问题, 本文提出抗肿瘤药物反应预测方法, 运用机器学习技术, 对患者肿瘤基因测序数据进行处理、特征提取及建模, 预测各种不同的抗肿瘤药物的疗效反应.首先, 提出基于多尺度关联规则的数据挖掘方法, 对基因组学数据进行不同尺度的特征挑选.进而通过累积窗函数对挑选后的基因组学数据进行局部累积, 进一步进行数据压缩, 提取具有较强整体表达性的基因特征信息.然后, 以多层全连接神经网络为模型、以提取的多尺度累积基因特征为输入样本, 进行训练和建模.最后, 分别采用特征融合和决策融合, 实现某一肿瘤基因测序数据对于各种不同抗肿瘤药物反应结果的预测.在COSMIC、GDSC数据库上的仿真实验表明, 本文方法在敏感性、特异性、准确率、特性曲线面积值等关键性能指标上均取得较优值.
针对抗肿瘤药物反应预测研究中的关键问题, 本文提出抗肿瘤药物反应预测方法, 运用机器学习技术, 对患者肿瘤基因测序数据进行处理、特征提取及建模, 实现对各种不同的抗肿瘤药物的疗效反应进行预测的方法.本文方法主要由数据预处理、多尺度局部累积特征提取、多层全连接神经网络和融合与决策四个模块组成, 具体结构框架如图1所示.
 | 图1 抗肿瘤药物反应预测方法框图Fig.1 Flow chart for prediction of antitumor drug response |
在数据预处理模块中, 本文对COSMIC、GDSC数据库提供的基因组学数据和药物反应数据进行筛选和数值化处理.在多尺度局部累积特征提取模块中, 首先采用多尺度关联规则挖掘方法对人体肿瘤细胞系的基因表达、基因突变和拷贝数变异数据进行特征挑选, 保留其中与药物反应存在关联的有用信息, 去除无关信息.然后, 在多尺度特征的基础上采用局部累积处理, 选择最优的局部累积特征观察窗提取每种尺度的局部累积特征.在多层全连接神经网络模块中, 以3个3层全连接神经网络为模型, 以提取的多尺度累积基因组学特征为输入样本, 分别进行训练和建模.在融合与决策模块中, 分别采用决策融合和特征融合, 设计具体的融合和决策算法, 实现某一肿瘤基因测序数据对于各种不同抗肿瘤药物反应结果的预测.
本文采用CCLP、GDSC数据库作为源数据.CCLP数据库提供1 001株人类肿瘤细胞系的基因表达、拷贝数变异和基因突变这3种常见的基因组学(特征个数大于60 000)的深入分析数据.GDSC数据库提供这1 001株肿瘤细胞系对265种抗肿瘤药物的药物反应测量值(如ln(IC50)值).为了不影响方法的预测性能, 本文选取其中的196种药物, 去除药物反应数据缺失比例大于20%的69种.
CCLP数据库提供的基因组学数据包括:16 445维基因表达谱、24 858维拷贝数变异和19 384维基因突变.文献[6]采用的预处理方法是将上述数据量化处理为符号表达的基因状态信息.为了与DLNNs进行公平对比, 本文采用与其相同的基因状态信息.同时依照文献[12]对上述基因状态的符号表达进行数值化处理, 以便后续的网络训练.具体为, 在基因表达状态中, 采用1表示基因的高表达, -1表示低表达, 0表示介于二者之间.在拷贝数变异状态中, 采用1表示超出重复片段正常值的基因, -1表示有缺失片段的基因, 0表示正常.在基因突变状态中, 采用1表示携带体细胞除沉默突变之外的所有单点突变, 其余突变类型用0表示.
在GDSC数据库中, 文献[6]采用零-均值规范化的ln(IC50)值划分预测网络的输出类别.其中, 阈值-1用于划分药物的敏感性(Sensitive)或不敏感性(Non-sensitive), 阈值1用于划分药物的耐药性(Resistant)或非耐药性(Non-Resistant).本文采用的理想标签是与对药物的敏感性、耐药性进行预测有关.如需对抗肿瘤药物的敏感性进行分类预测, 根据文献[6], 在训练过程中:对数据库上标有敏感的细胞系, 将标签值置为1, 作为正样本; 将其余未标有敏感的细胞系的标签值置为0, 作为负样本.如需对抗肿瘤药物的耐药性进行分类预测:对标有耐药的细胞系, 将标签值置为1, 作为正样本; 将其余未标有耐药的细胞系的标签值置为0, 作为负样本.
CCLP数据库提供超过6万维的基因组学数据, 如何从中提取合适的特征集并有效压缩特征维度是预测抗肿瘤药物反应的关键问题之一.文献[6]和文献[13]通过设定关联规则中一组确定的支持度阈值S0和可信度阈值C0, 采用Apriori算法[14]提取与药物反应密切关联的基因组学特征.这种在单一S0和C0下提取的基因组学特征称为单尺度特征.单尺度特征提取方法采用单一的特征挑选规则, 可能会遗漏原始基因组学数据中的某些有用信息.针对这一问题, 本文提出多尺度累积特征提取方法, 尽可能地从原始基因组学数据中提取更多有用的基因组学特征, 进一步提高基因特征的整体表达性.
1.3.1 多尺度关联规则挖掘
区别于单一的特征挑选规则(设定关联规则中单一的S0和C0), 通过分析文献[6]和文献[13], 本文发现设定多组S0和C0, 可提取不同尺度的基因组学特征.这相当于既细致又全面地分析原始的基因组学数据.采用多尺度关联规则挖掘方法提取多尺度基因组学特征的一般步骤可描述为:首先设定V种不同的强关联规则, 每种强关联规则均包含一组确定的支持度阈值Sv和可信度阈值Cv, v=1, 2, ···, V.在确定的Sv和Cv下, 采用Apriori算法从训练集T中挖掘NFv个形如“ 基因⇒ 某一种药物反应” 的强关联规则(满足支持度大于等于Sv, 可信度大于等于Cv的条件).提取NFv个强关联规则中的基因, 组成与某一抗肿瘤药物反应密切关联的一维基因组学特征Xv.当v≤ V时, 重复上述操作, 直到获得一组多尺度基因组学特征X=[X1, X2, ···, Xv].文献[6]设定S0=C0=0.004 48, 对应本文采用的训练样本数669, 其值为3/669.因此, 本文选择1/669、2/669和3/669这3种尺度的支持度阈值和可信度阈值, 用于提取多尺度基因组学特征, 即V=3.而0/669这一尺度相当于对原始基因组学数据未做任何特征提取, 本文并未采用.
对于尺度为v的基因组学特征数据集Xv={XEv, XMv, XCv}, 由基因表达特征XEv、基因突变特征XMv和拷贝数变异特征XCv组成.不同尺度下得到XEv、XMv和XCv的特征维度不同, 大小与特征提取时采用的S0和C0呈反比例关系.例如, 在较大的S0和C0下, 特征挑选规则要求“ 基因Gi高表达” 和“ 药物Dj的敏感性” 同时发生的概率较高.在此特征挑选规则下, 原始基因组学数据中大量与药物反应关联程度较低的基因被删除, 只保留重要的基因组学特征, 此时Xv的特征维度较低.而在较小的S0和C0下, 提取的基因组学特征会保留大量的原始基因组学数据, 此时Xv的特征维度较高.而以高维的基因组学特征为输入, 会造成后续预测子网络的处理难度增加.针对这一问题, 本文采用局部累积特征提取方法, 可进一步降低Xv的特征维度.
1.3.2 局部累积特征提取
为了进一步降低基因组学特征的维度, 缓解后续预测子网络的处理难度, 文献[6]采用从基因组学特征Xv中有放回地随机挑选一小部分特征的方法.此方法具有很强的随机性, 很难保证多次随机挑选的基因组学特征会全部覆盖Xv.此外, 由于人类基因组中通常只有小部分基因发生突变, 基因组学数据在很大程度上会呈现稀疏性.针对上述问题, 本文提出局部累积特征提取方法, 对每种尺度的基因组学特征进行以局部累积特征观察窗为单元的累积处理.设基因序列集为
{xk(n)|k=0, 1, ···, K-1, n=0, 1, ···, N-1},
其中, xk(n)为第k个基因序列, K为全部基因序列个数, N为基因序列长度.设Fk(i)为xk(n)的第i段累积特征, 则
Fk(i)=
其中, wM(m)为宽度为M的累积窗函数, LM为累积特征序列的长度.wM(m)的取值可根据同一种类型的基因组学数据内部的重要性确定.由于本文采用的基因组学数据是经量化后的基因状态信息, 该量化操作会损失基因组学数据内部的重要性信息, 因此, 本文同等对待在同种类型的基因组学数据中发生突变的基因, 将wM(m)取为如下矩形窗:
wM(m)=
对于提取基因突变序列的局部累积特征(记为FMk(i)), 将式(1)中的xk(n)采用相应的基因突变序列进行计算即可.在提取局部累积特征的过程中, 若最后一段包含的基因组学数据长度不足M, 对缺少的数据进行补0操作.此外, 考虑到基因表达状态包含1和-1两种非0情况, 为了防止累积运算中发生正负抵消的情况, 本文将基因表达序列(仍用xk(n)表示)分解成2个序列:
然后分别计算局部累积特征:
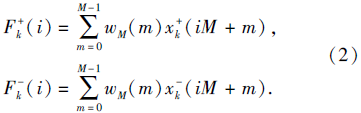
拷贝数变异序列与基因表达序列相同, 都是取值为1或-1的序列, 局部累积特征提取过程与基因表达序列一致, 仍按照式(2)求取局部累积特征.
从上述累积特征提取过程可看出, 若在特征观察窗范围内的一段基因组学特征中不存在发生突变的基因, 则这一段的累积特征输出为0.相反, 若存在发生突变的基因个数越多, 则这一段的累积特征输出会越大.这一结果与肿瘤的发生是由多个相关基因突变共同作用引起的思想吻合.局部累积特征提取的实质是在多尺度特征的基础上采用局部累积处理, 突出基因区域(或基因段)的整体影响, 而不是仅考虑个别基因位的影响, 从而可进一步提高特征的整体表达性.该方法不仅能反映肿瘤细胞系的特性, 还在保证基因组学特征全覆盖的同时降低特征维度和缓解稀疏性.
采用机器学习方法有效捕获生物标记物与抗肿瘤药物反应之间复杂的非线性关系是预测抗肿瘤药物反应的另一个关键问题, 现有研究方法已取得一定成果, 但也存在网络结构较复杂的情况[6, 9, 10].为了简化网络结构, 满足多尺度局部累积特征输入的需求, 进一步提升药物反应的预测性能, 本文设计一套抗肿瘤药物反应预测方法.该系统为每个单尺度的局部累积特征构建一个基于3层全连接神经网络的预测子网络, 并采用决策融合和特征融合, 分别融合3个子网络的预测输出或特征输出, 实现对抗肿瘤药物的敏感性或耐药性进行分类预测.
1.4.1 预测子网络的设计
针对肿瘤细胞系某一尺度的局部累积特征Fk, 本文基于3层全连接神经网络设计1个预测子网络, 用于预测某一种抗肿瘤药物对Fk的药物反应.多层全连接神经网络具有结构灵活的特点, 满足为每种药物设计适宜的网络结构的目的要求.本文设计的每个预测子网络均采用N0-N1-N2-N3的3层全连接神经网络结构, N1> N2, 其中, N0为输入特征维度, N1、N2为隐层节点个数, N3为输出节点个数.预测子网络中隐层的激活函数采用正切函数, 输出层的激活函数采用softmax.为了防止预测子网络过拟合, 每个隐层均使用失活层和L2正则化.
如果预测子网络是针对药物的敏感反应进行预测, 那么网络的两类输出节点分别表示药物作用在某一细胞系上表现为敏感或不敏感的可能性.对抗肿瘤药物反应的理想标签1和0进行One-Hot编码, 得到向量[1, 0]T或[0, 1]T作为预测子网络的理想输出.若药物作用在某一细胞系的药物反应越敏感, 预测其敏感性的输出越接近于1; 否则, 越接近于0.两类输出节点之和为1, 节点中取输出最大对应的类别作为预测子网络最终的预测结果.
1.4.2 损失函数的设计
分类问题中经常使用的损失函数是交叉熵损失函数, 显著特性是数量多的简单样本损失值会淹没数量少的困难样本损失值, 使网络模型的优化方向并不是预期所想.例如, 在目前已有的用于抗肿瘤药物反应研究的医学数据样本中, 肿瘤细胞系的用药反应表现为敏感或耐药的正样本个数通常是缺少的, 这样的数据分布会造成网络模型的预测性能尤其是正样本的预测性能下降.为了解决这一问题, 本文引入由Lin等[15]提出的改进的交叉熵损失函数Focal Loss, 核心思想是为正负样本分配不同的损失权重, 并在优化过程中逐渐降低简单样本的影响.Focal Loss优点是不需额外增加困难样本, 便可有效训练所有样本.Focal Loss的表达式为
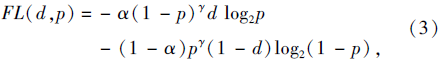
其中, d为训练数据中的某一细胞系(基因序列)对某种抗肿瘤药物表现为敏感(或不敏感)的理想标签(d=1表示敏感, d=0表示不敏感), p为所用方法对d的预测结果(pÎ [0, 1]).
由式(3)可看出, Focal Loss通过最小化交叉熵损失优化模型的训练过程.在交叉熵损失函数中:增加一个加权因子α , 平衡正负样本的比例; 增加一个调节因子(1-p)r, 调节简单样本损失降低的速率.α 和1-α 分别作为正、负样本的损失权重.随着γ 的增加, 调节因子能减小简单样本对损失的贡献, 扩大样本接收低损失的范围, 增加纠正错误分类样本的重要性.
1.4.3 融合与决策
不同尺度的基因组学特征包含不同的特征信息, 并对最终的预测结果起到不同的作用.本文针对如何合理有效地融合不同尺度这一问题进行研究.按照特征融合和网络预测的先后顺序, 分为早融合和晚融合.早融合是先融合多尺度特征, 再在融合后的特征上训练分类器.晚融合是对以不同尺度的特征为输入的预测子网络的输出结果进行融合.因此, 本文提出如下两种方法, 提高单尺度特征的抗肿瘤药物反应预测性能, 具体描述如下.
方法1为决策融合, 采用加权融合方法(Weighted Fusion, WF), 下文中均采用Proposed-WF表示该方法.加权融合方法是对以不同单尺度特征为输入的预测子网络的输出pv进行加权后, 得到多尺度特征对某种抗肿瘤药物反应的预测结果:
p=
最简单的wv取值为1/V, 即单尺度预测子网络的输出pv对最终药物反应预测结果p的贡献是平等的.此外, wv取值还可根据每个预测子网络采用训练集进行预测性能评估时得到的归一化AUC(Area under Curve)值或ACC(Accuracy)值等性能指标确定.此时, 单尺度预测子网络的输出pv对最终药物反应预测结果p的贡献与预测子网络的综合性能指标呈正比关系.
方法2为特征融合, 采用特征融合方法(Feature Fusion, FF), 下文中均采用Proposed-FF表示该方法.特征融合方法首先对以不同尺度局部累积特征为输入的网络N2层的输出特征进行融合, 再利用融合后的特征对某种抗肿瘤药物的敏感性或耐药性进行预测.Proposed-FF的一般步骤是级联每个预测子网络中N2层的特征输出yv(见文献[2]), 组成一个N'0维的多尺度特征[
本文的抗肿瘤药物反应预测方法在CPU+GPU平台上实现.实验平台配置为:Intel® CoreTM i7-7700@3.6 GHz, 内存32.00 GB, NVIDIA GeForce GTX 1080Ti.操作系统为Windows 7, 开发环境为Rstudio 1.1.453+Pycharm Edu 3.5.1+Keras框架.Apriori算法的实现使用R 3.4.0中的Arules 1.6.3程序包, 预测模块的实现使用Python3.5中的Keras 2.2.4库, 后端为TensorFlow1.4.0.
本文采用的评价指标为:敏感性(Sensitivity, SENS)、特异性(Specificity, SPEC)、约登指数(YD’ s Index), 全体样本准确率(ACC)和ROC(Receiver Operating Characteristic Curve)曲线下的面积AUC值.
为了定量计算上述性能评价指标, 根据各评价指标在抗肿瘤药物反应预测问题上的生物学含义, 在实验中首先分别计算出真阳性(True Positive, TP)、假阳性(False Positive, FP)、真阴性(True Negative, TN)、假阴性(False Negative, FN)4个实验测试统计值.其中, TP定义为:在对某种抗肿瘤药物反应进行预测的实验中, 预测为敏感的所有基因序列中其真实标签也为敏感的基因序列的个数.FP定义为:预测为敏感的所有基因序列中其真实标签为不敏感的基因序列的个数.TN定义为预测为不敏感的所有基因序列中其真实标签也为不敏感的基因序列的个数.FN定义为:预测为不敏感的所有基因序列中其真实标签为敏感的基因序列的个数.在此基础上分别计算SENS、SPEC、YD’ Index、ACC、AUC值:
SENS=
SPEC=
YD’ s Index = SENS+SPEC-1,
ACC=
AUC值指标是计算ROC特性曲线与横坐标轴围成的面积, 是与分类阈值无关, 对样本类别不平衡问题的鲁棒性的评价指标.
本文设计一套综合性的评估方案, 用于分析不同特征提取方法或不同网络结构对抗肿瘤药物反应预测性能的影响.方案的目的是合理评估本文方法在预测肿瘤细胞系对药物反应表现为敏感或耐药方面的性能.
为了公平对比, 本文与针对GDSC数据库中上百种药物进行抗肿瘤药物反应预测的方法进行对比分析, 具体包括:基线模型DLNNs, AutoBorutaRF[5]和MOLI[7].为了与DLNNs和MOLI进行公平对比, 本文采用相同的数据划分及预处理方式, 以基因表达、拷贝数变异和基因突变3种基因组学数据的状态信息作为细胞系的特征, 网络输出为分类肿瘤细胞系对抗肿瘤药物的敏感性或耐药性.DLNNs和MOLI的网络模型是依照文献[6]和文献[7]中提供的程序代码编程实现.AutoBorutaRF是以基因表达谱、拷贝数变异的实值和基因突变的状态信息3种基因组学数据作为细胞系的特征, 模型输出为分类肿瘤细胞系对抗肿瘤药物的敏感性.
为了避免本文方法过学习或欠学习, 实验时本文分别采用三折交叉验证和五折交叉验证.根据肿瘤细胞系的组织来源进行训练集和测试集的划分, 这是为了保证本文方法可学习到所有组织上的肿瘤细胞特征.具体以三折交叉验证为例, 在每次实验过程中, 将来自同一组织的肿瘤细胞系随机划分3份, 其中2份用于训练, 1份用于测试.该过程重复3次, 每次留不同的部分作为测试集, 最终的预测性能指标是3次测试的平均结果.若某种组织只包含2个细胞系, 训练集和测试集均分.若某种组织只包含1个细胞系, 分配到训练集中.经上述划分, 训练集共包含669个细胞系样本, 测试集共包含332个细胞系样本, 比例接近2:1.
各方法的预测性能对比如表1所示, 表中黑体数字表示最优值.AutoBorutaRF针对GDSC数据库上98种药物, 经十折交叉验证后得到平均预测性能.DLNNs、MOLI及本文方法是针对GDSC数据库上196种药物, 经三折交叉验证后得到平均预测性能.
| 表1 各方法的预测性能对比 Table 1 Prediction performance comparison of different methods |
由表1可知, 本文方法在全部性能指标上均有所提升.由于在GDSC数据库上存在正负样本不平衡问题, 模型学习到正样本的特征不足, 造成敏感性指标值较低.DLNNs的敏感性指标值明显低于特异性指标值也说明这一点.为了克服正负样本不平衡问题, AutoBorutaRF采用如下策略:将负样本分成T份, 每份负样本Ti的样本数近似等于正样本R.每份Ti和R训练一个RF(Random Forest)分类器.根据每个RF投票获得最终的抗肿瘤药物反应预测结果.由表1可看出, AutoBorutaRF的敏感性指标值高于DLNNs.Proposed-WF和Proposed-FF采用Focal Loss损失函数解决这一问题, 其中, Focal Loss的参数设置为γ =2, α =0.2.Proposed-WF和Proposed-FF得到的敏感性指标值均超过对比方法, 比次优的AutoBorutaRF分别提高0.101和0.097.Proposed-WF和Proposed-FF得到的ACC指标值比次优的MOLI分别提高0.056和0.059, AUC指标值比次优的MOLI分别提高0.138和0.136.
在GDSC数据库的196种抗肿瘤药物中, 采用Proposed-WF和Proposed-FF进行预测, 获得AUC值最高的药物是达拉菲尼(Dabrafenib), 分别为0.970和0.964.达拉菲尼用于治疗转移性黑色素瘤, 是BRAF基因突变的抑制剂.本文采用的多尺度累积特征提取方法在挑选基因组学特征的过程中, 证实达拉菲尼可能会对BRAF基因产生重要的影响.因此, BRAF基因也作为预测达拉菲尼药物反应的输入特征.
2.3.1 不同基因组学特征对预测性能的影响
为了分析不同特征提取方法对抗肿瘤药物反应预测性能的影响, 本节在药物反应预测模型基本相似的前提下, 开展随机挑选特征、单尺度特征、单尺度局部累积特征、多尺度特征和多尺度局部累积特征的分解实验.实验中随机挑选特征是指文献[6]的特征提取方法.3组单尺度基因组学特征X1、X2、X3是通过分别设置3条关联规则中的最小支持度阈值和最小可信度阈值为1/669、2/669和3/669得到的.单尺度局部累积特征是指采用累积窗函数从单尺度基因组学特征Xv中提取的局部累积特征Fv.本文采用7种不同窗宽的累积窗函数, 分别对输入特征X1、X2和X3提取局部累积特征, 即M=1, 5, 10, 20, 30, 40, 50.实验发现:相比其它M, 单尺度特征X3在M=10下提取的局部累积特征F3的药物反应预测性能相对较优, 而X2和X1在M=30下提取的F2和F1的预测性能相对较优.因此, 在多尺度局部累积特征融合方法中, 仅采用这些预测性能相对较优的单尺度局部累积特征进行特征融合.多尺度特征MX是指采用Proposed-FF分别将单尺度基因组学特征X1、X2、X3的N2层特征输出后级联得到的特征.多尺度局部累积特征MF是指采用Proposed-FF分别将单尺度局部累积特征F1、F2、F3的N2层特征输出后级联得到的特征.经五折交叉验证后, 本文统计上述不同的特征提取方法对抗肿瘤药物反应预测的性能, 结果如表2所示, 表中黑体数字表示最优值.
| 表2 不同基因组学特征的预测性能对比 Table 2 Prediction performance comparison of different genomic features |
由表2可知, 采用以多尺度特征MX为输入, 对抗肿瘤药物的敏感性或耐药性进行分类预测的性能优于采用单尺度特征X1、X2或X3.同样, 采用多尺度局部累积特征MF的预测性能也优于采用单尺度局部累积特征F1, F2或F3.例如, 采用多尺度局部累积特征MF进行预测, 得到最优性能的ACC值为0.835, AUC值为0.888, 相比相对较优的采用单尺度局部累积特征F1, ACC值和AUC值分别提高0.005和0.005.这说明相比单尺度特征, 多尺度特征提取方法能提取更多对药物反应预测性能有帮助的基因组学特征, 进一步提高每种单尺度特征的抗肿瘤药物反应预测性能.此外, 采用局部累积特征对药物反应进行分类预测的各项性能指标值均优于不采用局部累积特征的.例如, 相比不采用局部累积的多尺度特征MX, 采用多尺度局部累积特征MF进行预测得到的ACC值和AUC值, 分别提高0.065和0.122.这说明考虑多个相关基因的共同作用, 在特征观察窗内提取基因组学局部区域的累积特征, 可进一步提高基因组学特征的整体表达性.
2.3.2 不同网络结构对预测性能的影响
为了分析不同网络结构对抗肿瘤药物反应预测性能的影响, 本文基于多层全连接神经网络, 设计4种网络结构的分解实验.研究GDSC数据库中的196种抗肿瘤药物, 每种药物作用在肿瘤细胞系上的药物反应分别表现为敏感或耐药两种情况.因此, 每种药物反应采用1个预测器进行预测, 共需要392个预测器.实验中网络1采用N0-N1-N2-N3的3层结构, 该结构也是目前本文所有实验中采用的网络结构.本文尝试修改网络结构中的隐层节点数N1和N2, 统计在不同网络结构下的药物反应预测性能.经大量实验后发现, 当N1=
为了融合不同尺度的特征, Proposed-FF采用的网络结构为N'0=30和N'1=8.网络2和网络3均采用N0-N1-N2-N3-N4的4层结构.网络2的N1~N3节点数大小相同, 取值均为4
N1=4
网络4采用N0-N1-N2-N3-N4-N5-N6的6层结构, N1~N6节点数大小先增大后减小, 取值分别为
N1=N5=
经五折交叉验证后, 本文统计网络1~网络4的抗肿瘤药物反应预测性能, 结果如表3所示, 表中黑体数字表示最优值.
| 表3 不同网络结构的预测性能对比 Table 3 Prediction performance comparison of different network structures |
由表3可看出, 虽然随着神经网络的层数增加, 药物反应预测性能在特异性指标上有一定程度的提升, 但敏感性指标却下降明显.网络4的特异性指标为0.896, 相比网络1, 提升0.054; 敏感性指标为0.650, 相比网络1, 下降0.115.此外, 由于医学数据的正负样本不平衡问题, 当样本量占比较大的负样本的正确率提升时, 全体样本的准确率也随之提升.采用网络4的ACC值为0.864, 相比网络1, 提升0.029.但此时另一关键的综合性能指标AUC值却随着神经网络的层数增加而有不同程度的下降.例如, 本文采用的3层的神经网络1获得的AUC值为0.888, 相比采用6层的神经网络4, 提升0.013.从网络模型的参数量角度分析, 网络4的参数量为
20N0+(N0+12)
网络1的参数量为
(4N0+44)
例如, 当N0=100时, 网络4的参数量为3 122, 网络1的参数量为872.这说明采用更多层数的神经网络需要更多的计算代价, 却未得到更好的预测性能.在综合权衡考虑下, 本文决定采用网络1的3层神经网络结构.
有研究指出:AUC值在0.5~0.7时, 模型的预测准确性较低.AUC值在0.7~0.9时, 模型具有较好的预测准确性.AUC值大于0.9时, 模型具有较高的预测准确性[16].AutoBorutaRF也采用AUC≥ 0.7作为合格的模型预测性能评判标准.本文统计当前研究方法中AUC≥ 0.7的比例, 与Proposed-WF和Proposed-FF进行对比.AutoBorutaRF中AUC≥ 0.7的比例是参考该文给出的实验结果, 统计GDSC数据库上98种药物获得的, 而DLNNs和MOLI是统计GDSC数据库上196种药物获得的.经三折交叉验证后, 本文统计上述不同方法获得预测性能AUC≥ 0.7的比例, 具体如下:AutoBorutaRF为60.2%, MOLI为70.4%, DLNNs为72.0%, Proposed-WF为100%, Proposed-FF为100%.由仿真结果可看出, 本文方法所有抗肿瘤药物反应预测模型的AUC值均大于0.7, 这表明基于多尺度局部累积特征和多层全连接神经网络的抗肿瘤药物反应预测模型具有较优的预测准确性, 均是合格的预测模型.以Proposed-FF为例, 具体AUC值比例分布如下:AUC值在0.7~0.8的比例为0.3%, AUC值在0.8~0.9的比例为76.4%, AUC值在0.9~1的比例为23.3%.
抗肿瘤药物反应预测的目的是要面向临床应用, 针对不同患者, 通过该模型提供个性化治疗的药物推荐.在预测准确性方面, 全球疾病误诊率高达30%, 即ACC≥ 0.7.由于肿瘤存在异质性, 医生的评估准确率在实际中更低.经三折交叉验证后, 本文方法与AutoBorutaRF、DLNNs、MOLI进行对比, 各方法获得预测性能ACC≥ 0.7的比例如下:DLNNs为73.5%, AutoBorutaRF为84.8%, MOLI为86.5%, Proposed-WF为100%, Proposed-FF为100%.Auto-BorutaRF中ACC≥ 0.7的比例是参考该文给出的实验结果, 统计GDSC数据库上98种药物获得的, 而DLNNs和MOLI是统计GDSC数据库上196种药物获得的.
本文方法所有抗肿瘤药物反应预测模型的ACC值均大于0.7, 具体ACC值比例分布如下:ACC值在0.7~0.8的比例为17.99%, ACC值在0.8~0.9的比例为80.16%, ACC值在0.9~1的比例为1.85%.因此, 本文方法有利于确定药物与肿瘤细胞系基因组学特征之间的关系, 有望在个性化医疗领域发挥重要作用.
本文结合现有公开发布的人类肿瘤细胞系的基因组学数据, 提出基于多尺度局部累积特征和神经网络的抗肿瘤药物反应预测方法.采用多尺度关联规则挖掘方法提取基因组学数据的多尺度特征, 避免由于采用单一尺度特征提取时可能会遗漏某些有用信息而存在的问题.提出多尺度局部累积特征提取方法, 突出基因区域(或基因段)的整体影响, 而不是仅考虑个别基因位的影响, 进一步提高特征的整体表达性.提出一套由3个3层子网络, 并且分别通过采用决策融合或特征融合方法进行融合的预测模型, 结构简单、计算量较小、性能稳定、泛化性较好.本文工作是在COSMIC、GDSC数据库提供的1 001株细胞系的基因组学数据和196种抗肿瘤药物反应数据的基础上进行的.大量实验表明, 本文方法在敏感性、特异性、准确率及AUC值等关键性能指标上均较优.
随着基因组学研究的发展, 还将有更多的基因组数据出现, 这为进一步采用深度卷积神经网络模型进行抗肿瘤药物反应预测提供研究基础, 也是作者下一步的主要研究方向之一.此外, 文献[17]的研究表明沉默突变也有可能会导致人类癌症, 因此如何利用沉默突变信息进行抗肿瘤药物反应的预测也是一个值得研究的方向.
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|