

{kind=link}
{kind=link}
{kind=link}
基于BERT和CNN的致病剪接突变预测方法
[宋程程1  , 赵依然
, 赵依然1 , 李晓艳1 , 夏俊峰1 ]
, 赵依然, 李晓艳, 夏俊峰]
|
|



作者简介:

宋程程,硕士研究生,主要研究方向为生物信息学、深度学习.E-mail:1484311435@qq.com.

赵依然,硕士研究生,主要研究方向为生物信息学.E-mail:18811212175@163.com.

李晓艳,博士,讲师,主要研究方向为生物信息学.E-mail:lixiaoyan@ahu.edu.cn.
遗传诊断中的一个关键挑战是评估与剪接相关的致病遗传突变.现有致病剪接突变预测工具大多基于传统的机器学习方法,主要依赖人工提取的剪接特征,从而限制预测性能的提升,尤其对于非经典剪接突变,性能较差.因此,文中提出基于BERT(Bidirectional Encoder Representations from Transformers)和CNN(Convolutional Neural Network)的致病剪接突变预测方法(BERT and CNN-Based Deleterious Splicing Mutation Prediction Method, BCsplice).BCsplice中BERT模块可全面提取序列的上下文信息,与提取局部特征的CNN结合后,可充分学习序列的语义信息,预测剪接突变致病性.非经典剪接突变的影响往往更依赖序列上下文的深层语义信息,通过CNN将BERT的多级别语义信息进行组合提取,可获得丰富的信息表示,有助于识别非经典剪接突变.对比实验表明BCsplice性能较优,尤其是在非经典剪接区表现出一定性能优势,有助于识别致病剪接突变和临床遗传诊断.
About Author:
SONG Chengcheng, Master student. Her research interests include bioinformatics and deep learning. ZHAO Yiran, Master student. Her research interests include bioinformatics.
LI Xiaoyan, Ph.D., lecturer. Her research interests include bioinformatics.
A key challenge in genetic diagnosis is the assessment of pathogenic genetic mutations related to splicing. Existing predictive tools for pathogenic splicing mutations are mostly based on traditional machine learning methods, heavily relying on manually extracted splicing features. Thereby the predictive performance is limited, especially for non-canonical splicing mutation producing poor performance. Therefore, a bidirectional encoder representations from transformers(BERT) and convolutional neural network(CNN)-based deleterious splicing mutation prediction method(BCsplice) is proposed. The BERT module in BCsplice comprehensively extracts contextual information of sequences. While combined with CNN that extracts local features, BERT module can adequately learn the semantic information of sequences and predict the pathogenicity of splicing mutations. The impact of non-canonical splicing mutations often relies more on deep semantic information of sequence context. By combining and extracting the multi-level semantic information of BERT through CNN, rich information representations can be obtained, aiding in the identification of non-canonical splicing mutations. Comparative experiments demonstrate the superior performance of BCsplice, especially exhibiting certain performance advantages in non-canonical splicing regions, and it contributes to the identification of pathogenic splicing mutations and clinical genetic diagnosis.
剪接是真核生物一种重要的转录后调控过程, 通过切除内含子和连接外显子可提高RNA(Ribo-nucleic Acid)和蛋白质水平的多样性和复杂性[1, 2, 3].多项研究表明, 90%以上具有多个外显子的基因会进行可变剪接[4, 5].但剪接过程可能受到基因组序列变异的影响或破坏, 导致原本的剪接位点失去功能或通过破坏剪接体组装以改变剪接过程[6, 7, 8, 9].这种剪接异常可能会引起蛋白质的合成异常, 进而导致疾病发生[10, 11].
研究发现影响剪接的突变与多种疾病密切相关[12, 13, 14, 15].然而, 目前还没有通用的规则解释剪接突变的致病性.美国医学遗传学与基因组学会(Ameri-can College of Medical Genetics and Genomics, ACMG)在指南中指出, 在功能丧失的已知机制的基因中, 位于经典剪接位点(外显子和内含子边界的GT与AG二核苷酸位置)上的突变往往具有致病性[16].对于非经典剪接突变, 由于该区域无明显的保守序列特征, 且影响剪接的方式多种多样[17, 18], 确定其致病性仍然具有挑战性[19].因此, 迫切需要开发剪接突变致病性预测算法, 尤其是针对非经典剪接突变的算法, 促进在下一代疾病诊断测序中有效地对致病剪接突变进行优先排序.
现有的广谱性致病突变预测方法可预测包含剪接突变在内的多种突变的致病性, 但由于该类方法纳入的剪接生物学特征有限, 限制其对剪接突变致病性的预测[20, 21].预测剪接分子效应的工具也可对剪接突变致病性进行预测.例如:SpliceAI[22]是一个深度残差神经网络, 可通过野生型和突变序列得分的差异预测剪接突变的致病性.类似的预测剪接分子效应的工具还有很多, 如Maximum Entropy Mo-dels[23]、MMsplice(Modular Modeling of Splicing)[24]、CADD-Splice[25]、SPiP(Splicing Prediction Pipeline)[26]等.然而, 预测剪接的分子效应与预测剪接突变致病性是两个截然不同的任务.
目前, 针对剪接突变致病性预测, 学者们已经提出多种方法.S-CAP(Splicing Clinically Applicable Pathogenicity Prediction)[27]使用梯度增强树分类器构建模型, 定义与剪接位点相关的6个区域, 包括外显子及其边界50 bp, 用于预测影响RNA剪接的遗传突变的致病性.SQUIRLS(Super Quick Information-Content Random-Forest Learning of Splice Variants)[19]为剪接供体和受体训练两个随机森林分类器, 并通过逻辑回归组合它们的输出, 得出最终的剪接突变致病性预测分数.
上述预测致病剪接突变的方法大多基于传统机器学习的方法, 依赖人工提取的剪接生物学特征.这种依赖性可能会使某些剪接突变数据在预测过程中被忽略, 导致方法预测的不够全面.同时, 由于缺乏足够的致病剪接突变正样本数据, 现有深度学习工具中的传统神经网络模型无法充分发挥优势.此外, 非经典剪接突变往往难以识别, 现有预测工具对非经典剪接突变的预测精度仍有待提高.因此, 亟需提出一种致病剪接突变预测方法, 对剪接突变功能效应进行优先排序, 改善疾病的诊断和治疗.
序列编码内部存在多义性和遥远的语义关系, 这是自然语言的关键属性.以往的研究表明, DNA(Deoxyribonucieic Acid)序列与人类语言具有极大的相似性[28], 也存在一词多义的现象.例如:相同的顺式调控元件(Cis-Regulatory Elements, CREs)在不同的上下游核苷酸序列中往往具有不同功能; 间隔较远的多个CREs可能会协同, 对启动子产生不同的功能作用.遗传突变可能会通过影响CREs导致异常剪接[29, 30].由此可见, 剪接突变的功能后果与其上下游序列密切相关, 尤其是那些发生在多义性的序列片段中的突变.
因此, 本文提出基于BERT(Bidirectional Enco-der Representations from Transformer)[31]和CNN(Con-volutional Neural Networks)[32]的致病剪接突变预测方法(BERT and CNN-Based Deleterious Splicing Mu-tation Prediction Method, BCsplice).BCsplice中BERT模块可全面提取序列的上下文信息, 与提取局部特征的CNN结合后, 可充分学习序列的语义信息, 预测剪接突变致病性.非经典剪接突变的影响往往更依赖序列上下文的深层语义信息, 通过CNN将BERT的多级别语义信息进行组合提取, 可获得丰富的信息表示, 有助于识别非经典剪接突变.对比实验表明BCsplice性能较优, 尤其是在非经典剪接区表现出一定性能优势, 有助于识别致病剪接突变和临床遗传诊断.
基于Transformers 编码器的语言模型BERT引入预训练和微调的范式, 在许多自然语言处理任务中取得较优性能.BERT首先从大量的无标签数据中发展通用的理解, 再通过在特定任务上的微调取得较优性能, 即使使用少量的数据也能获得出色的表现.目前相关研究将BERT基于DNA序列进行建模, 称为DNABERT[33].因此, 本文采用DNA-BERT的预训练模型作为BCsplice中提取序列上下文信息的一部分.此外, 在使用BERT处理分类任务时, 通常会使用BERT最后一层的信息.然而, Devlin等[31]对比BERT不同层的组合发现, 相比顶层的输出, 最后四个隐藏层的输出组合可编码更多的信息.因此, 本文选择提取BERT最后四个隐层的向量, 并进一步进行组合处理.考虑到CNN在提取局部特征方面具有显著优势, 以及经典的TextCNN在文本分类领域中的良好表现, 将BERT最后四个隐层的输出与TextCNN结合, 用于特征提取.
因此本文提出基于BERT和CNN的致病剪接突变预测方法(BCsplice), 整体架构如图1所示.BCsplice首先处理数据集, 再输入序列.由于遗传突变在不同的上下游核苷酸序列中可能会产生不同的功能后果, 突变位点的语义变化与上下文信息密切相关.因此, 首先提取突变位点的上下游基因组序列数据, 并将突变前后的基因组序列转换为一组使用k-mer标记表示的序列作为模型的输入.序列中还包括一个[CLS]标记(表示整个句子含义的标签)和两个[SEP]标记(表示句子分隔符, 在突变前后的序列结尾分别添加).输入序列首先经过嵌入层, 再送入BERT模块.然后, 将BERT最后四个隐层的[CLS]向量输出进行拼接, 拼接后的向量通过TextCNN进行卷积处理.最后, 通过一个分类层输出剪接突变是否致病.
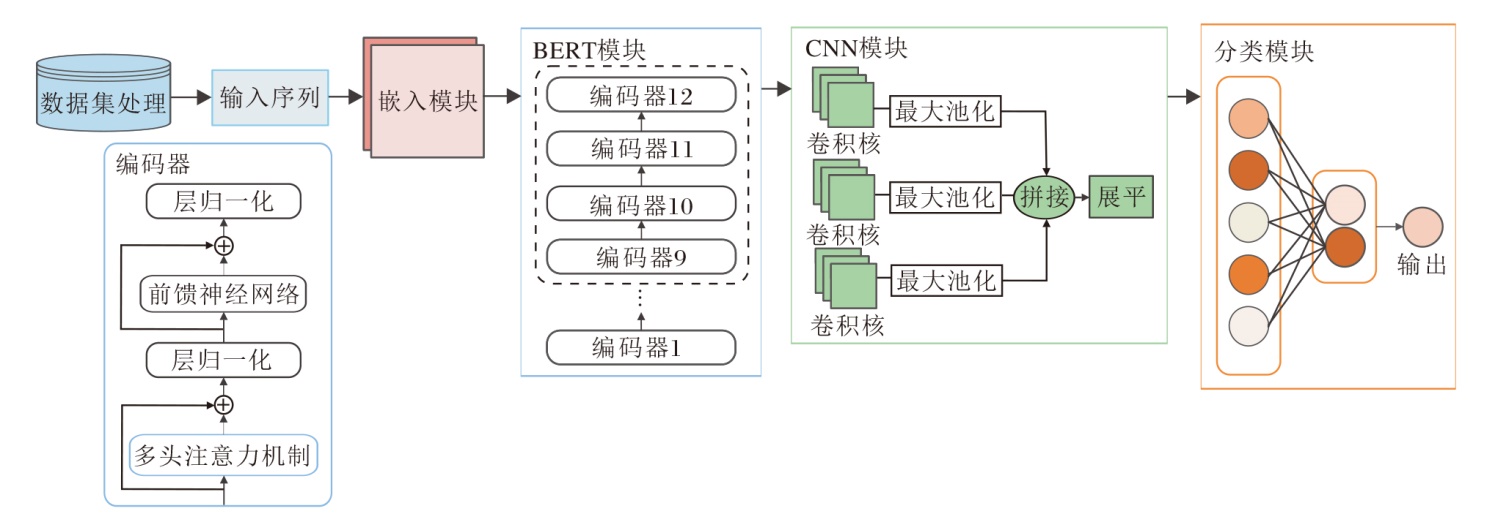 | 图1 BCsplice框架图Fig.1 Architecture of BCsplice |
BCsplice主要包括4个模块:嵌入模块、BERT模块、CNN模块、分类模块.各模块的细节描述如下.
1)嵌入模块.输入序列通过k-mer标记表示后, 每k个碱基相当于一个词(这里以k=6为例, 即每6个碱基作为一个词).嵌入模块包含Token Embe-dding与Positional Embedding.Token Embedding将每个词转换为768维的向量, 作为词的向量表示.Positional Embedding对序列中每个词的位置信息进行编码, 使模型能学习输入序列中每个词的顺序属性.最后将每个词对应位置的Token Embedding与Positional Embedding相加, 得到嵌入向量矩阵.
2)BERT模块.BERT的主要结构是Transformer中的编码器单元, 这里共有12层编码器.每个编码器单元依次由多头注意力机制、层归一化、前馈神经网络和层归一化叠加产生.
3)CNN模块.使用3个相同大小的卷积核对BERT的输出矩阵进行卷积操作.卷积核的大小设置为2× 768.卷积操作提取输入矩阵中的局部特征.卷积后得到的特征向量通过ReLU激活函数进行非线性映射, 增强表示能力.然后, 通过一个最大池化层对特征向量进行降维, 选择每个卷积核输出的最大值作为该卷积核的池化输出.最后, 将3个池化输出进行拼接, 形成一个更丰富的特征表示.
4)分类模块.通过一个线性层预测输入序列是否致病, 得到最终的输出结果.
为了构建一个高质量的与剪接突变相关(Splicing-Associated Variant, SAV)的数据集, 首先整合人类基因突变数据库(Human Gene Mutation Database, HGMD专业版2022.4)[34]上与剪接相关的突变数据, 共得到34 852条剪接突变数据, 其中包含突变类型为splice、exonic-splice、canonical-splice的数据.同时移除在Y染色体上的唯一一条剪接突变数据, 最终仅保留染色体1~22和X上的突变数据.然后, 根据人类参考基因组版本为GRCh37和致病性标签为DM(Disease Causing Mutation), 对剪接突变数据进行筛选, 最终获得26 550条致病剪接突变数据.另外, 还从ClinVar(https://www.ncbi.nlm.nih.gov/clinvar, 发布于2022年7月9日的VCF文件)[35]中获取402 763条临床意义为Benign的突变数据.
由于本文的致病剪接突变数据仅包含单核苷酸突变, 因此进一步筛选突变类型为单核苷酸的良性突变数据(GRCh37版本).同时, 为了保证负样本数据的置信度较高, 选择ReviewStatus为criteria provi-ded, multiple submitters, no conflicts的突变数据, 并限定染色体为1~22和X.
经过上述筛选, 最终得到32 064条良性突变数据.根据文献[27], 将剪接区域定义为外显子及其周围50 bp, 并筛选位于剪接区的突变数据, 从而获得30 772条良性剪接突变数据.
致病剪接突变与良性剪接突变数据的来源不同, 导致存在少量数据标签矛盾.因此, 移除正负样本标签不一致的数据, 最终得到26 543条致病剪接突变数据和30 765条良性剪接突变数据.另外, 鉴于非经典剪接突变的临床意义较难解释, 根据剪接区域的不同(如表1所示), 进一步将剪接突变数据划分到经典剪接区(Canonical Splicing Region)与非经典剪接区(Non-Canonical Splicing Region).
为了减少来自同一基因的不同突变造成的偏差, 采用Cytogenetic Band划分训练集和测试集.首先, 从总共721个染色体条带中随机选择20%(144个)的染色体条带, 将这些条带中包含的突变作为测试集.然后, 将剩下的80%(577个)染色体条带中包含的突变作为训练集.染色体条带是指突变位点所处的染色体区段, 每个突变都与特定的染色体区段关联.该方法的目的是减少位于同一基因中的不同突变同时被用于训练和测试的几率, 因为相邻的突变可能具有相似的特征, 可能导致结果偏差[19].通过Cytogenetic Band, 可确保同一染色体区段内的突变要么用于训练, 要么用于测试, 而不会同时用于训练和测试.划分后得到包含45 999条数据的训练集和包含11 309条数据的测试集, 具体如表2所示.
| 表2 数据集划分情况 Table 2 Division of dataset |
本文使用PyTorch框架构建BCsplice.模型以DNABERT为基础架构, 采用其默认参数, 详细参数见表3.对于输入的k-mer序列, 文献[35]研究表明, 当k=6时, 模型具有最优性能, 因此设置k=6, 即每6个碱基作为一个词.
| 表3 BCsplice参数信息 Table 3 Parameters of BCsplice |
本文使用7个常用指标评估BCsplice分别在全基因组区域和非经典剪接区的性能表现.具体指标如下:Accuracy(ACC)、ROC(Receiver Operating Cha-racteristic Curve)曲线下面积(Area under the ROC Curve, AUC), 精确率-召回率曲线下面积(Area under the Precision-Recall Curve, AUPR)、F1 score(F1)、马修斯相关系数(Matthews Correlation Coeffi-cient, MCC)、精确率(Precision)、召回率(Recall).这些评价指标是根据混淆矩阵计算的, 参数包括真正例(True Positive, TP)、真反例(True Negative, TN)、假正例(False Positive, FP)和假反例(False Nega-tive, FN), 具体计算方式定义如下:
$\begin{array}{l} A C C=\frac{T P+T N}{T P+F P+T N+F N}, \\ F 1=2\left(\frac{\text { Precision } \cdot \text { Recall }}{\text { Precision }+ \text { Recall }}\right), \\ M C C= \\ \frac{T P \cdot T N-F P \cdot F N}{\sqrt{(T P+F P)(T P+F N)(T N+F P)(T N+F N)}}, \\ \text { Precision }=\frac{T P}{T P+F P}, \\ \text { Recall }=\frac{T P}{T P+F N} .\end{array}$
一般来说, 各指标值越高, 模型性能越优.在进行性能对比时, 主要考虑AUC与AUPR, 因为在这7个评价指标中, AUC与AUPR是最重要的2个综合评价指标.
经典剪接区的突变位于外显子与内含子边界± 2 bp位置, 具有明显的基序特征和位置特征, 通常容易解释.非经典剪接区的突变相对更隐蔽, 致病性较难确定.因此, 本文将突变分为2个区域:经典剪接区和非经典剪接区.外显子和内含子边界的GT与AG二核苷酸位置的突变是发生在经典剪接区的突变, 这个位置的突变绝大多数都是致病的[16].在本文设计的数据集上, 经典剪接区的致病突变有21 965条, 而良性突变只有25条.由此可见, 经典剪接区上的良性突变极少, 仅占0.08%, 良性突变基本上都位于非经典剪接区.此外, 根据本文的分类结果, 经典剪接区的正负样本非常不平衡.尽管非经典剪接区的正负样本也存在不平衡, 但相比之下, 在非经典剪接区发生的突变并不像经典剪接区那样直观, 其致病性需要进一步研究.基于不同区域的致病性情况和突变数据的分布情况, 在接下来的性能对比中主要关注非经典剪接区和全基因组区域(包括经典剪接区和非经典剪接区).测试集在非经典剪接区和全基因组区域上的具体分布情况如表4所示.
| 表4 测试集在不同剪接区的分布情况 Table 4 Distribution of test set in different splicing regions |
为了研究BCsplice中BERT的隐层向量经过TextCNN处理后对模型性能提升的重要性, 定义BERT-TextCNN和BERT, 进行针对TextCNN模块的消融实验.由于经典剪接突变通常会对剪接产生有害影响, 故预测非经典剪接突变的致病性更具挑战性.因此, 除了在全基因组区域进行分析以外, 还对非经典剪接区进行单独分析.在列出的评价指标中, AUC与AUPR是最重要的两个综合评价指标, 因此主要根据这两个指标进行性能对比.
在全基因组区域, 首先对模型进行五折交叉验证和测试数据集上的性能评估, 具体如表5所示.根据表中结果可见, BERT-TextCNN在各个评价指标上都有性能提升.而在非经典剪接区域, BERT-TextCNN和BERT的指标值对比如表6所示.由表可见, BERT-TextCNN性能提升更显著, BERT-Text-CNN的AUC值为0.947, AUPR值为0.828, 相比BERT分别提高5.1%和15.2%.
| 表5 各模型在五折交叉验证和测试集上的性能 Table 5 Performance of different models on 5-fold cross-validation and test set |
| 表6 各模型在非经典剪接区测试集上的性能 Table 6 Performance of models on test set of non-canonical splicing region |
每个编码层的句子向量表示不同级别的语义信息.浅层的编码层能更好地捕捉低级别的语义信息, 而深层的编码层能更好地捕捉高级别的语义信息.通过TextCNN将多个编码层的语义信息进行组合提取, 既能获取原始的全局语义信息, 又能获得序列信息中关键的特征.这可能是模型性能提升的重要原因之一.此外, 非经典剪接区突变的影响更隐晦, 对于序列上下文的深层语义信息更依赖.
因此, 提取多级别的隐含信息对于预测非经典剪接突变的致病性具有重要意义.
为了进一步验证BCsplice的优越性, 选择如下剪接突变预测方法进行对比:SQUIRLS[19]、Spli-ceAI[22]、CADD-Splice[25]、SPiP[26]、S-CAP[27]、MLC-splice(Machine Learning-Based Classification of Splice Sites Variants)[36].
SpliceAI是一种可从pre-mRNA转录本序列中预测剪接连接(Splice Junctions)的方法, 通过对比突变前后序列的分数差异以预测突变的致病性.CADD-splice整合其它工具的剪接分数, 并优化对剪接突变效应的预测.MLCsplice是一个集成的机器学习方法, 用于预测突变对剪接的影响.SPiP也是一种机器学习工具, 用于检测外显子和内含子突变对mRNA剪接的影响.
为了对比方法的性能, 使用11 309条测试数据, 并在全基因组区域和非经典剪接区域进行实验, 结果如表7和表8所示.
| 表7 各方法在全基因组区上的性能对比 Table 7 Performance comparison of different methods in whole genome region |
| 表8 各方法在非经典剪接区上的性能对比 Table 8 Performance comparison of different methods in non-canonical splicing region |
在本文的测试数据中, 并不是每个工具都能对所有突变给出预测结果.因此将对比预测工具分为两类:1)不能对所有突变给出预测结果的工具, 即存在缺失突变(CADD-splice、SpliceAI、S-CAP、MLC-splice); 2)可对所有测试突变给出预测结果, 即没有缺失突变(SPiP、SQUIRLS).在表中使用“ 类别” 进行标注以区分这两类工具.需要特别注意的是, BCsplice也属于没有缺失突变的这一类工具.因此BCsplice与SPiP、SQUIRLS可在同一水平上进行对比.
在全基因区域, 使用由5 319个致病剪接突变和5 990个良性突变组成的测试集.根据表7的评估结果显示, BCsplice在AUC和AUPR指标上取得最高值(AUC=0.987, AUPR=0.988).相比存在缺失突变的工具, BC-splice表现出明显的优势.BCsplice能对所有突变数据进行预测, 而其余4种方法都存在一定的缺失突变.具体缺失突变的数量见表9.MLCsplice缺失的突变数量最多, 达到10 092条(10092÷ 11309≈ 89%).其次是S-CAP, 缺失5 076条突变(5076÷ 11309≈ 45%).相比没有缺失突变的工具, BCsplice在AUC和AUPR值上的提升约为1%~2%(见图2), 也显示出一定的优势.
| 表9 各方法在全基因组区上的混淆矩阵值 Table 9 Confusion matrix values of different methods in whole genome region |
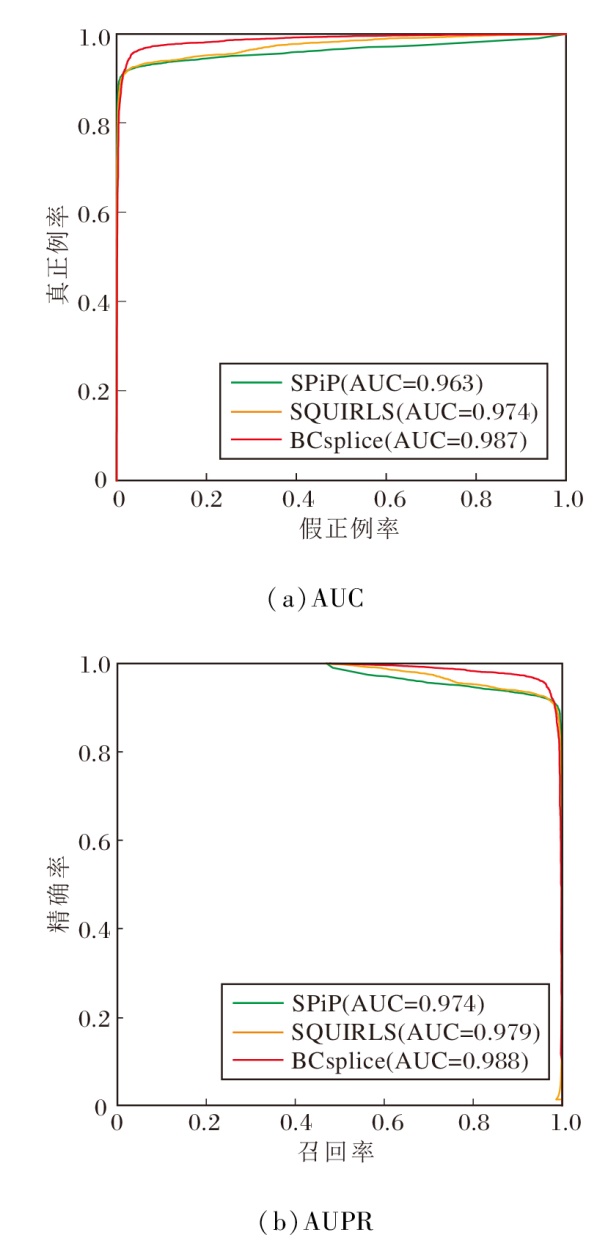 | 图2 各方法在全基因组区域上的综合性能对比Fig.2 Comprehensive performance comparison of different methods in whole genome region |
综合来看, 相比对比方法, BCsplice性能更优或相当.这是因为对于使用者来说, 一个理想的方法不仅需要高精确度, 还需要能分析更广泛范围的突变.BCsplice可对全基因组上任何区域的突变进行预测, 而当前的预测工具大多存在剪接突变预测分数缺失的情况.那些能对所有剪接突变进行预测的工具, 在性能上并未高于BCsplice, 特别是在非经典剪接区域中.
另外需要说明的是, 由于获取MLCsplice和S-CAP的预测分数需要先计算其它多种工具的特征分数, 过程较耗时, 因此本文直接使用预先计算好的MLCsplice和S-CAP的人类基因组突变分数, 这可能是两个预测工具缺失值较多的原因.对于CADD-Splice, 本文使用线上预测功能, 下载包含PHRED标注突变致病性评分的tsv文件.为了评估SpliceAI, 根据其文献中提供的网站资源, 在服务器上下载SpliceAI工具包, SpliceAI为每个突变提供4个Δ 分数, 这4个分数中的最大值代表该突变的Δ Score.然而, 对于CADD-Splice和SpliceAI, 目前无法明确它们存在缺失值的具体原因.可能是由于它们依赖于某些剪接特征或受到基因组区域的限制, 导致无法预测所有的剪接突变.对于所有对比方法, 在本文的测试集上, 使用find_optimal_cutoff函数生成具有高AUC的最佳阈值, 确保性能对比的准确性与一致性.
非经典剪接突变的临床意义较难解释, 发生在非经典剪接区的突变也难以识别.大量的预测方法专注于经典剪接区, 对非经典剪接区突变的预测能力较弱.因此本文在非经典剪接区上也进行性能对比, 具体如表10所示.在本文的测试集上, 非经典剪接区的致病剪接突变有916条, 良性突变有5 985条.
| 表10 各方法在非经典剪接区上的混淆矩阵值 Table 10 Confusion matrix values of different methods in non-canonical splicing region |
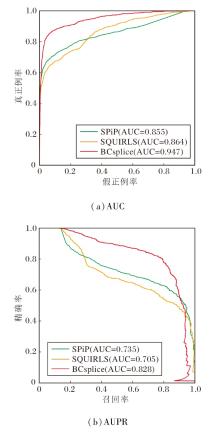
由表10可见, 相比预测区域在全基因组区域, 所有方法在非经典剪接区上都显示出一定程度的性能下降.其中CADD-Splice、SPiP与SQUIRLS性能下降最明显, 这可能与非经典剪接区的样本数据不平衡有关.在非经典剪接区域, 本文依旧将对比方法分为两类.相比存在缺失突变的方法, MLCsplice在AUC和AUPR指标上具有最高值.然而, 在本文的测试集上, MLCsplice存在5 994个突变的缺失情况(占总数的87%), 具体缺失情况见表10.相比没有缺失突变的方法, BCsplice具有显著优势(AUC=0.947, AUPR=0.828).相比SPiP(AUC=0.855, AUPR=0.735)与SQUIRLS(AUC=0.864, AUPR=0.705), BCsplice的综合性能提升8%~12%, 具体如图3所示.
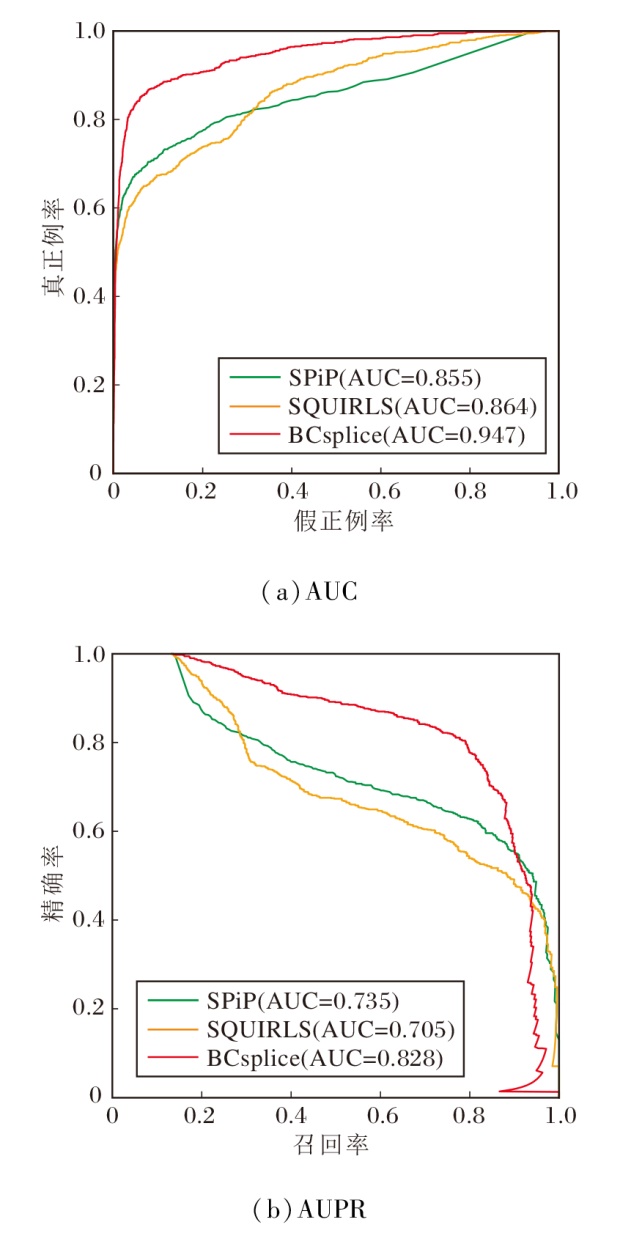 | 图3 各方法在非经典剪接区上的综合性能对比Fig.3 Comprehensive performance comparison of different methods in non-canonical splicing region |
因此, BCsplice在不同情况下都表现出独特的优势.非经典剪接突变通常更依赖序列上下文的深层语义信息.BCsplice通过结合CNN和BERT以捕捉多级别的语义信息, 这可能是其性能较优的原因之一.
本文提出基于BERT和CNN的致病剪接突变预测方法(BCsplice), 用于预测剪接突变的致病性.BCsplice利用基因组序列数据全面捕捉序列上下文信息, 鉴别有害的剪接突变.该方法显示自身的优越性与实用性, 尤其是对于非经典剪接突变的预测, 可支持改善和扩展对剪接突变的诊断能力, 并对突变产生的剪接影响进行评估, 为识别剪接突变做出一定的贡献.此外, BCsplice是一个深度学习算法, 通过自动学习序列信息以提取特征, 但是这些特征并不是容易理解的有明确生物学含义的特征.今后的研究方向之一是进一步探索方法的可解释性, 并对突变导致的剪接缺陷进行解释说明, 这有助于理解剪接突变如何导致人类疾病, 并更深入了解剪接这一精密过程的调节机制.
本文责任编委 周水庚
Recommended by Associate Editor ZHOU Shuigeng
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
|